|
|
|
|
multidivtime (multidistribute とされていることもある) は,Bayes 法を用いて,塩基およびアミノ酸配列に基づいて分岐年代推定を行うプログラムです.ノースカロライナ州立大学の Jeffrey Thorne さんが作成しました.ここでは解析例として multidivtime と一緒に配布されている Gene1, Gene2, Gene3 というデータを使用します. フローチャート (pdf) を作成したので,参照してください.このページでは Mac 上で Unix を操作して multidivtime を動かし,塩基配列データを解析しています.アミノ酸配列の解析は こちらをご覧下さい.
multidivtime でバグを経験したことは,ほとんどありません.最初は大変ですが,慣れると PAUP などの user friendly なプログラムより使いやすい気さえします.
正しい読み方というものがあるのかわかりませんが,私の所属する研究グループでは,multidivtime のことを「マルタイディブタイム」と呼んでいます.
|
|
最近の年代推定解析
Idaho で行われた Evolution 2009 に参加してきました.multidivtime を用いた解析は一つも見かけませんでした.アメリカの研究者のほとんどは,BEAST を使って年代推定の解析をしていました.使い慣れると BEAST もいいそうです.しかしマニュアルがないので,研究グループ内で情報を交換して解析を進めているそうです.私の周辺では,BEAST はあまり評判が良くないです (2009 年 6 月).
mcmctree の登場
残念なことに,制作者である Thorne さんは multidivtime の version up を行っていないようです.このため私としては,multidivtime のアイデアを十分に取り入れた mcmctree をこれからは使った方が良いように思えます.ただ,mcmctree は,化石制約の事前分布に用いるパラメータ設定など,未だ開発途中です.しばらくは multidivtime と mcmctree の結果を比較してみる必要がありそうです (2009 年 6 月)
AC coding 解析
multidivtime に関わらず,AC coding (G→A, T→C と変換し,transition を無効にする解析) に対応しないプログラムがたくさんあります.少し荒技ですが,わずか数十塩基程度を変換しないでおくと (G と T を残しておく),解析が走り出すことが多いです.外群など一つの OTU だけで私は行っています.おそらく,尤度を計算するときに塩基組成に 0 が入っていると,解析が止まってしまうのでしょう.同様の処置は,mtREV (mtMAM だったかも知れません) のマトリックスなどで見られます.アミノ酸置換速度が 0 と推定された要素を 0.1 などに置き換えています.ちなみに解析ソフトの作者には AC (あるいは RY) 解析を好まない人もいます.
Taxon sampling を密にすると推定年代が古くなる?
multidivtime だけの問題ではないようで,Linder et al. (2005) でいくつかのプログラムにおいて taxon sampling の粗密がどのように推定値に影響するか比較しています.また Yang and Yoder (2004) では,密にとりすぎるよりも適切な数の OTU に絞った方が妥当な推定値が得られるはず,としており,multidivtime の推定方法に基づいてその理由を述べています.
話はそれますが Yoder and Yang (2004) の Fig. 4 では,それほど generation をとらなくても推定値が収束に向かうことを示しています.
Linder HP, Hardy CR, Rutschmann F
Taxon sampling effects in molecular clock dating: An example from the African Restionaceae
MOLECULAR PHYLOGENETICS AND EVOLUTION 35 (3): 569-582 JUN 2005
Yoder AD, Yang ZH
Divergence dates for Malagasy lemurs estimated from multiple gene loci: geological and evolutionary context
MOLECULAR ECOLOGY 13 (4): 757-773 APR 2004
解析可能な OTU 数を増やす
multidivtime は 200 OTU (種,あるいは個体) の解析まで可能なようにこのページに書いてしまっていましたが,どうも 100 OTU がデフォルトの限界のようです.これ以上の OTU を用いる場合は,コンパイルする前に multidivtime のソースコードを書き換える必要があります.estbranches は書き換えなくても問題なく動くようです.
multidivtime.c の 49 行目にある MAXNODE を書き換えます.
#define MAXNODE 200 /* maximum number of nodes on rooted tree */
ここで注意したいのが,MAXNODE はどうも分岐ではなく枝の数を指している (らしい) 点です (node というと分岐点を指していると考えていましたが,multidivtime では全体を通して,node という表現に注意する必要があります).MAXNODE は末端の枝と内部枝も含めた枝 (番号が付く全ての枝) のことのようで,例えば 130 OTU からなる二分岐 tree (有根樹) であれば 257 (2N-3) 個の枝があることになります.少し多めに設定しておいた方が良いかも知れません.
この情報は宮正樹先生に教えて頂きました.
Rutschmann さんのマニュアルにある F.A.Q. #6 にもこの件に関して説明がありますのでご覧下さい
mtREV + F + &Gamma モデル
mtREV + F + &Gamma モデルを用いたアミノ酸解析のやり方を図にしてみました.
こちらをご覧下さい.
メカニズムの理解に挑戦
2006 年 11 月 15 日に,
「Phylomethods Fall 2006 Dating - r8s, multidivtime (PDF file)」
という題名でゼミをしました.メカニズムの理解に挑戦していますので,ご覧ください.
|
|
| 1.1. 必要なもの |
- Mac (ターミナルを使います).ここでは OS 10.4.3 を用いています.ターミナルは Dock に入れておくと便利です. Windows の場合はコマンドプロンプトで操作します.
- multidivtime (http://statgen.ncsu.edu/thorne/multidivtime.html)
- PAML package (http://abacus.gene.ucl.ac.uk/software/paml.html)
- テキストエディタ (Mac なら BBEdit か mi)
*重要事項: 全ての infile は改行コードを Unix にして保存します.プログラムがうまく走らないときは,まず改行コードを疑ってみます.余談ですが,BBEdit は値段もアカデミックバージョンなら $50 程度と比較的安いうえに,とても使いやすいエディターだと思います.コンピューターサイエンティストの間では定番になっているようです.
- 配列データ (塩基 or アミノ酸配列.ここでは testseq.Gene1 など).
- TreeView .Version 0.4.1* が良いです.Tree をドローソフトにコピー & ペースとして,各枝を分解できるので,Fig. を簡単に作成できます.
- 系統樹.あらかじめ推定しておきます.ここでは tree.Gene1 や example.tree などのファイルです.得られる推定値は,用いる樹形の違いにかなり影響されると思います.私は試したことがありませんが,多分岐を含んでも解析はできるみたいです.MacClade で tree を .phylip 形式で保存すると,OTU 名のあとにスペースが入ってしまいますが,問題なく解析できます.
- ルートノード (外群と内群を結ぶ点) の年代.得られる内群の分岐年代は,個々の枝に設定した較正点よりも,ルートノードの年代に大きく影響されます.この値は慎重に設定する必要があります.
- 系統的位置が明確になっている化石の年代.脊椎動物であれば,まずは The Fossil Records, vol 2 (Benton, 1993) を参照するといいと思います.2007 年になって,化石学者である Benton さんが実験動物の年代推定に有用な化石情報と分岐制約を考察しています (Benton and Donoghue, 2007).地質年代に関しては,Gradstein et al. (2005) を引用する文献が増えています.彼らの website から地質年代表 (PDF) をダウンロードすれば,実際の年代 (Mya) を調べられます.
文献に関しては,こちらをご覧下さい.
- Developer tools.すぐ下のコラム「1.2. Developer tools のインストール」 を参照して下さい.
|
1.2. X code (Developer tool) のインストール
|
| アプリケーションフォルダに入っている App Store を使って,X code をダウンロード&インストールします.Mac の OS が Snow leopard ぐらいになってから,X code のインストール方法が App Store を使ってやるようになっています.Apple ID が必要です. |
 |
App Store で X code をインストールしただけでは,cc などのコンパイラは使えるようになりません.アプリケーションフォルダにある Xcode というアプリケーションを立ち上げて,Preference > Downloads にある Command Line Tools を選んで Install を押します.詳しくはこちらをご覧ください.
Mac OS Mountain Lion で multidistribute のコンパイルを試したところ,cc だとエラーメッセージが出ましたが,一応コンパイルされたプログラムは現れます.以下のように gcc を用いると,エラーメッセージは出ませんでした.
gcc -DDNA estbranches.c -DHESSIAN arrayutl.c inv3.c -lm -O -o estbranches
(2012 年 7 月).
|
1.3. PAML のコンパイル
|
- PAML の OSX バージョンをダウンロードします.
Windows バージョンはコンパイルせずにそのまま使えます.Windows バージョンは,解凍すると bin というフォルダにアプリケーション (.exe という拡張子がついています) が入っています.
- ダウンロードした paml3.14b.OSX_G5.tar をダブルクリックし,解凍します.
Mac の CPU が G4 や G3 の場合は,古いバージョンを解凍して試して下さい.
- 解凍してできた paml3.14 をホームディレクトリー (私であれば,inoue という名前のディレクトリーになっている) に移します.
- ターミナルを開きます.
- ターミナルに
ls
と入力すると,inoue フォルダーの内部にあるファイルが確認できます.
- cd paml3.14
と入力し,paml3.14 フォルダーの内部に入ります.
- cd src
と入力し,src フォルダーの内部に入ります.
- make -f Makefile.UNIX
と入力し,コンパイルを行います.詳しくは readme ファイルを参照して下さい.Mac の性能にもよると思いますが.5 分ぐらいかかるでしょうか.
- さらに
cp baseml basemlg codeml evolver pamp yn00 mcmctree chi2 ..
で上位のディレクトリ (「..」が上部ディレクトリを表します) にコンパイルされた baseml, basemlg,.... といった 8 つのアプリケーションを移動します.cp というコマンドはコピーを意味するので,src ディレクトリ (今自分がいるディレクトリ) の上部ディレクトリにもプログラムがコピーされます.実際には,プログラムを mv コマンドや,あるいは単純にファインダー上の操作でドラッグ&ドロップすることでプログラムをコピー&ペーストして使うこともできます.余談ですが,プログラムの名前を変えても正常に動作するので,ソースコードを変更してコンパイルした場合にちがう名前のプログラム (ex. codeml_F) にする事も可能です.
- rm *.o
と入力し,不要になった「.o」ファイルを消去する.
- ファインダー上 (通常のドラッグアンドドロップ) で,baseml を multidivtime フォルダーにコピー&ペーストします.
|
1.4. multidivtime のコンパイル
|
multidivtime をダウンロードし,ホームディレクトリー (私であれば,inoue という名前のディレクトリーになっている) に移します.実際には desktop などにディレクトリーを作成しても大丈夫ですが,浅い階層に作成した方が扱いが便利です.「multidivtime の操作に少し慣れてきたから,今書いている論文と同じディレクトリーで解析したい」という場合には,ドラッグ&ドロップで実は簡単に cd (移動) できます.ご存じのない方はこちらをご覧下さい.
ダウンロードしてきたフォルダー「multidistribute」には以下 3 種類のアプリケーションが入っています.
paml2modelinf
baseml で推定された gamma や Ts/Tv などのパラメーターが書かれた outputfile を estbranches で読めるファイルに書き換える.
estbranches
シーケンスと各種パラメーターに基づいてい,樹長と樹長の分散・共分散行列を算出する.樹長と樹長の分散・共分散行列は尤度を概算するのに用いているようです (Thorne and Kishino, 2005).
multidivtime
estbranches の outputfile と分岐に施した時間制約に基づき,ベイズ法を用いて分岐年代を推定する.ここではシーケンスが用いられていません.
|
|
- ターミナルを開きます.
- ターミナルに
ls
と入力すると,inoue フォルダーの内部にあるファイルが確認できます.Unix に入力するコマンドは,readme ファイルからコピーして使うと間違えにくくて良いです.
- cd multidistribute
と入力し,multidistribute フォルダーの内部に入ります.
- readme を開きます.
- estbranches をコンパイルします.
塩基配列の場合は,
cc -DDNA estbranches.c -DHESSIAN arrayutl.c inv3.c -lm -O -o estbranches
をコピーし,ターミナルに入力します.
アミノ酸配列の場合は,
cc estbranches.c -DHESSIAN arrayutl.c inv3.c -lm -O -o estbranches
と入力します.
- paml2modelinf (baseml の output を estbranches の infile に書き換えるプログラム) をコンパイルします.
cc paml2modelinf.c arrayutl.c -lm -O -o paml2modelinf
をコピーし,ターミナルに入力します.
- multidivtime をコンパイルします.
cc multidivtime.c inv3.c arrayutl.c -lm -O -o multidivtime
をコピーし,ターミナルに入力します.
- readme ファイルによると,以上のコンパイルで,「-O」(数字の 0 ではなく大文字の O) となっている部分を「-O3 」や「-O4」にしたほうが高速化されるそうです.
- 一度コンパイルしてしまえば,multidistribute の各種アプリケーションは,ドラッグ & ロップでフォルダー間を移動しても問題なく作動します.このため新しい解析を行う場合は,フォルダーを作成し,必要なアプリケーションとファイルをコピー & ペーストすれば良いです.
|
1.5. シーケンスファイル
|
testseq.Gene1 を使います.MacClade を用いて Phylip 形式 (sequential: OTU 名の後にすべての配列が折り返されずに記載される方式) で保存すれば作成できます.以下は自分のデータで infile を作るときの注意点です.
MrBayes の解析に用いた Nexus 形式のファイルから,各パーティションを書き出すには PAUP を用います.詳しくはこちらのページにある「データパーティションの書き出し」をご覧下さい.
|
- mi の検索・置換機能では,リターンなどの変換も可能です (もちろん,free ウェアでなければ BBEdit でもできます).正規表現一覧はこちら.ただし,multidivtime の解析には関係ありませんが,T3 のシーケンスファイルには「>」があり,これには注意を要します.私の知っている限りでは,mi や TeraPad (Win) の「>」は不都合で,T3 を動かすには Mega で「>」を同じ文字に変換する必要があります.
- BBEdit には,複数のファイルを選択して一括しての検索置換を行う機能があります.これを用いると複数の partition (遺伝子) のファイルを同時に作成できます.
- N や - はすべて ? に変換するようにマニュアルには指示されています.しかし - はそのままでも問題なく解析が走ります.
- OTU の名前はおそらく 10 文字以内です.OTU の文字数はそろえておきます.特殊な文字は使わない方がよいと思います.「_」は使用できますが避けた方がよいと思います (MacClade で Phylip 形式に保存すると,スペースが _ に変換されてしまうので,注意してください).もちろん tree ファイルと OTU 名を統一して下さい.
- 最初の OTU と同じ配列を示す「.」(ドット) は認識されないようです.
- 改行コードは Unix にしておきます.
- 複数の遺伝子や座位からなるデータセットを解析する場合は,別々のファイルを作成します.この場合も OTU 名は統一して下さい.データセットごとに OTU 数が異なっていても (例: Gene1 は 5 つ,Gene2 は 7 つ),解析は可能なようです.
- Windows バージョンでは,最後の OTU の配列の後に,リターンが入っていない場合は,estbranches_aa.exe がうまく動きませんでした.
- ファイルの最後の改行は複数回にした方が無難です.詳しくはこちらをご覧下さい.
- 極端に短い配列では estbranches の解析が途中でストップすることがあります.とくにアミノ酸の解析で生じやすい問題だと思います.
私の経験では (38OTU, 54 残基),あるアミノ酸がデータにまったく含まれていなかったため,codeml によって推定された各種マトリクスに 0 が含まれてしまい,estbranches の解析が始まりはするものの終わらなくなってしまいました.この場合は,他の遺伝子で推定したモデルを estbranches の解析に転用するなどの措置が必要になると思います.
|
1.6. tree ファイル
|
最良と思われる系統樹を予め用意しておきます.Gene1.tree および example.tree を参照して下さい.MacClade を用いて Phylip 形式で保存することで作成できます.解析がうまく走らない原因の多くが tree ファイルの不備です.tree ファイルを作成するときの注意点を書いておきます.
|
- 外群 (通常は 1〜3 種程度でしょうか) は単系統になるようにします.以前は 1 種しか外群として設定できないと思っていましたが,最近 2 種を設定して問題なく解析することができました.
- 根幹の分岐は 2 分岐かあるいは多分岐にします.しかし baseml では 2 分岐にした場合,自動的に多分岐にされるか,あるいは解析がストップしてしまうので,根幹の分岐は多分岐にしておいた方が無難だと思います.
- 5 1
(Taxon_A,((Taxon_B,Taxon_C),(Taxon_D,Taxon_E)),Outgroup);
のように,最初の行は OTU 数と系統樹の数 (この行は何が書いてあっても良いらしいです) を書きます.系統樹の記載では,外群を最も右側に置きます.
- OTU 名に含まれるスペースは MacClade で _ にされますが,PAUP ではそのままスペースになってしまい,シーケンスファイルと tree ファイルで名前が一致しないトラブルがあったので注意して下さい.
- TreeView を用いて,tree ファイルに誤りがないか確認すると良いです.MacClade 4.08 で tree ファイルを Phylip 形式で保存すると,なぜか tree の先頭にスペースが一文字入ってしまいます.スペースを取り除かないと TreeView はうまく動きません.
- 多分岐が含まれていても,解析はできるようです.
|
|
baseml を使います.ここでは F84 + Γ (5 category) モデルが選択されています.
|
Input:
testseq.Gene1
シーケンスファイル
Gene1.tree
tree ファイル
5 1
((Taxon_A,(Taxon_C,(Taxon_D,Taxon_E))),Outgroup);
Gene1.ctl (塩基配列の場合)
コントロールファイル.モデルなどを設定
aga2aa.ctl (アミノ酸配列の場合)
Output:
Gene1.out
進化モデルのパラメーター推定値
oGene1
スクリーン出力
- ./baseml Gene1.ctl > oGene1
と入力します.
*注意: Unix の世界ではカレントディレクトリを示す「./」がないとアプリケーションを起動できません.
- Gene2 と Gene3 も同じように解析を行います.
./baseml Gene2.ctl > oGene2
./baseml Gene3.ctl > oGene3
- アミノ酸の解析で,ガンマ補正などを行わず,mtmam などのモデルを用いる場合は,パラメーターを推定する必要はありません.ダウンロードしたフォルダーにある既存の「evalfl.jtt」や「evecfl.jtt」というファイルを使うことになります.もしアミノ酸の解析でガンマ補正やアミノ酸の頻度の補正を行いたい場合は,multidivtime の HP にある熊澤先生が書いた記事を参考にして下さい.
- 進化モデルは F84 かあるいは HKY 85 が使えます.multidivtime ではこれ以上パラメータの多いモデルを扱っていません.
塩基組成は不均一: 最尤的に推定 (nho = 1).
Ts/Tv (κ):最尤的に推定 (fix_kappa = 0).
座位間の進化速度は不均一で離散ガンマモデルを適用: ガンマの形状パラメーターαは推定 (fix_alpha = 0),速度のカテゴリーは 5 つ (ncatG = 5).
- baseml (paml) の ver 3.15 で 3rd コドンポジションを解析していたところ,
「distances out of range: diff= 110.275938」
というエラーが出て,中途半端な outfile しか得られませんでした.どうも distance が大きすぎると動かないようです.baseml を ver. 3.14 にしたら問題なく作動しました.この問題に限らず,どうも ver. 3.14 と 3.15 では大きな変更があったようで,他の解析でもうまく作動しないことがありました.
|
| 3. baseml の outfile を estbranches の infile に変換 |
|
|
paml2modelinf を使います.baseml の outfile をみて modelinf.Gene1 を作成することも可能です.
|
Input:
Gene1.out
進化モデルのパラメーター
Output:
modelinf.Gene1
書き換えられた進化モデルのパラメーター
- ./paml2modelinf Gene1.out modelinf.Gene1
と入力.
*参考: paml2modelinf は,baseml の出力ファイルである Gene1.out を estbranches の入力ファイルとなる modelinf.Gene1 に書き換えるプログラムです.paml2modelinf を 用いないで baseml の結果をみて,modelinf.Gene1 を手作業で書き換えることも可能です.
- Gene2 と Gene3 も同じように解析を行います.
- gamma 補正をしない設定で得た baseml の出力では,paml2modelinf がうまく作動しませんでした.F84 で 3 カテゴリーの gamma 補正ではうまく作動しました.paml2modeinf がうまく動かない場合は,プログラムと一緒に配布されている modelinf ファイルを参考にしてファイルを手動で書き換える必要があります.それほど難しくはないです.
|
|
|
estbranches を使います.baseml で推定されたパラメーターの値を用いて,系統樹の樹長,分散・共分散行列 (variance-covariance matrix) を最尤推定します.
|
Input:
modelinf.Gene1
書き換えられた進化モデルのパラメーター.
testseq
testseq.Gene1 をコピーし,testseq という名前に変更.estbranches は testseq という名前のシーケンスファイルを自動的に読み込みます.ファイルの最後の改行が 1 回しか入っていないと,解析が停止することがあるようです.最後の改行は複数回にしておいた方が無難なようです.
Gene1.tree
tree ファイル.
5 1
((Taxon_A,(Taxon_C,(Taxon_D,Taxon_E))),Outgroup);
hmmcntrl.dat
コントロールファイル.
Output:
oest.Gene1
tree,樹長,分散・共分散が記載されています.
out.oest.Gene1
スクリーン出力.
- ./estbranches oest.Gene1 > out.oest.Gene1
と入力.このままではアプリケーションが実行されているかわからないので,まず最初に
> out.oest.Gene1
の部分を除いて実行してみて下さい.ダイレクトに結果がスクリーンにあらわれるので,アプリケーションが正常に作動しているかどうかがわかります.ここでエラーメッセージが出なくなったら,上記のようにアプリケーションを実行して下さい.
- Gene2 と Gene3 も同じように解析を行います.
hmmcntrl.dat ファイル内部の Gene1 部分をそれぞれ Gene2,Gene3 と書き換えることを忘れないで下さい.もちろん testseq ファイルも新たに作り直します.
|
|
- grep lnL Gene*.out
と入力し,baseml で推定された尤度を出力します.
- grep 'FINAL LIKE' out.oes*
と入力し,estbranches で推定された尤度を出力します.それぞれのパーティションで同じような値が得られていれば良いです.
- 「grep」は引数に記された文字が含まれているファイルを拾い出し,その部分だけを表示するコマンドです.
|
|
|
multidivtime を用いて MCMC 解析を行います. multidivtime では,推定された樹長の多変量正規分布を用いて尤度関数が近似されるので,シーケンスデータも進化モデルも用いていません.
|
Input:
oest.Gene1, oest.Gene2, oest.Gene3,
tree,樹長,分散・共分散行列.
example.tree
Taxon B がない以外 (Gene2 と 3 にはある) は,Gene1.tree と実際には同じです.
junk on this line
((Taxon_A,((Taxon_B,Taxon_C),(Taxon_D,Taxon_E))),Outgroup);
multicntrl.dat (DNA の場合)
multicntrl.dat (アミノ酸の場合)
コントロールファイル.MCMC パラメーター.較正点の設定など.パラメーターの設定や英文表現などに関しては Teeling et al. (2003) が少しは参考になると思います.
inseed
シード番号.確認のため同じ解析 2 回以上にわたって行う場合は,inseed を異なる数字にする必要があります.
Output:
out.example
分岐時間と速度の事後平均などをの情報を要約したもの.
tree.example
時間軸付き系統樹.この tree file を TreeView で開き樹長を反映した tree にして,図の作成に用います.
ratio.example
MCMC 法によってサンプルされたパラメーターの値.
node.example
事後確率.このファイルの最後に,各分岐について推定された年代が書かれています.推定年代の 95% 信頼区間もここにあります.Excel と Illustrator などの表作成機能などをうまく使うことで,時間軸付き系統樹に 95% 信頼区間を入れることができます.
samp.Gene1
MCMC のサンプルが入っているようです.私はまだやったことがありませんが,R (アール) を使うことで,尤度の収束 (convergence) を見ることができるようです.
- 解析を行うに当たり,tree に化石情報から得られる constraint を設定する必要があります.まず以下のコマンドを入力し,ノード番号を設定します.
./multidivtime numbers
- ターミナル上にノード番号のついた Newik 形式の系統樹が表示されます.これをエディターの新規ファイルにコピー&ペーストし,保存したファイルを TreeView で開いてノード番号を確認します.TreeView に持っていく前に,検索置換で : (コロン) を取り除く必要があります. root node の番号だけが出ませんが,これは単なるバグだと思います.得られた node numbert 付きの系統樹に化石情報などを当てはめて,各 node の制約を考えます.制約の記入方法は multicntrl.dat をご覧下さい.
upper constraint の設定についてこちらで少し考察してみました.
- MCMC 解析をスタートします.
./multidivtime example > out.example &
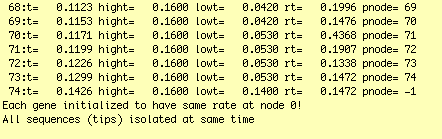
inseed 内部の数字を変更して,少なくとも 2 回解析を行い,結果が一定しているかどうか確認します.まずは「> out.example &」を外して「./multidivtime example」としてコマンドを入力し,解析がうまく走るかどうか確認してください.うまく行けば以下のような表示になります.「control + C」で解析を中断できます.
|
|
こちらをご覧下さい.
|
|
|
|
Thorne JL. 2003. http://statgen.ncsu.edu/thorne/multidivtime.html
ダウンロードしたフォルダにある readme ファイルに,解析を行うにあたり必要なことがほぼすべて書いてあります.
Rutschmann F. Bayesian molecular dating using PAML/multidivtime: A step-by-step manual, version 1.5 (July 2005).
ftp://statgen.ncsu.edu/pub/thorne/bayesiandating1.5.pdf
英語ですが,使い方が分かり易く説明されています.
|
|
|
このマニュアルは,他の研究者から寄せて頂いた情報も参考にして作成しています.
お気づきの点があれば,ご連絡ください.
情報を提供して下さった皆さま,ありがとうございます.
|
|