|
|
|
|
mcmctree はベイズ法に基づいて分岐年代推定を行うソフトウェアです.PAML に含まれています.multidistribute と比べてみると,分岐の制約に softbounds (事後確率が必ずしも事前確率の範囲内でない制約) が採用されている点と,枝ごとの rate を独立させて推定することができる点などが異なります.使い勝手などを考えても multidistribute に取って代わるプログラムと言えます.
当初 mcmctree は尤度を概算しない Exact calculation だけを採用していましが,解析速度が非常に遅く,10 OTU を超えるような解析は実用的なスピードではありませんでした.paml4 になってから,multidistribute と同様に概算する Approximate likelihood calculation が採用され,解析速度の弱点が改善されました.mcmctree を使う場合は,ぜひ PAML を最新版にアップグレードして下さい.
|
|
|
|
|
- コントロールファイルないでのコメントアウト
paml ではコメントアウトに「*」が用いられています.このため,コントロールファイルにある以下のような行 (* の右側) は,コメントアウトされ読み飛ばされてしまいます.
* finetune = 1: 0.06 0.5 0.008 0.12 0.4 * times, rates, mixing, paras,
- FigTree.tre ファイル
PAML45 から,MCMC アルゴリズムによって time tree が推定された後,FigTree.tre ファイルが作成されるようになれました.これによって,信頼区間付きの time tree が描けるようになりました [2012 年 1 月].
- Fine tune の自動化
(Version 45 からうまく動くようになったようです.2012 年 1 月)
Version 44e から fineture を自動的に調節するオプションがつきました.ただ,これはまだ十分な検討はなされていないようです.mcmctree.ctl ファイルにある finetue に,数字とコロンが加わりました.0 は手動で調節,1 は自動調節みたいです.詳しくはマニュアルをご覧下さい.同じファイルを古い version で動かすとおかしな acceptance proportion が得られるので注意してください [2011 年 7 月].
finetune = 0: 0.04 0.2 0.3 0.1 0.3
- Hard/soft bound の選択
Version 44c あたりから,Hard bound と Soft bound の設定を,tree file に書き込めるようになりました.詳しくは公式マニュアルか,以下にある「Hard/Soft bound 設定」をご覧下さい (2011 年 7 月).
- GTR モデル
mcmctree マニュアルにはまだ書いていないようですが,塩基配列の解析で GTR モデルを選ぶこともできます.model = 7 です.マニュアル baseml の箇所を参照してください [2010 年 9 月].
- mcmctree と baseml は同じバージョンを使いましょう.
異なるバージョンで推定した in.BV ファイル (枝長と分散・共分散マトリクスが書かれている) をつかって mcmctree でベイズ解析を行うと,おかしな結果が得られることがあるそうです.推定年代や尤度が巨大な値になっていました [2010 年 6 月].
- 年代推定の解析速度
どのソフトウェアでも言えることだと思いますが,年代推定は OTU の数によって解析速度が大きく左右されます.配列の長さも多少は影響します.ただ,tree search のように OTU が多くなると解析速度が劇的に遅くなる,ということはないです.Tree search は OTU が増えると得られる tree の数が劇的に増えるのに対し,年代推定では分岐の数 (time, rate, 枝長の分散・共分散パラメーター) が増えるだけだからだと思います (2009 年 6 月).
- コーシー分布
コーシー分布は,下限制約を施す際に化石記録の信頼度を反映させるのに適した分布と考えられ,version 4.2 に組み込まれました.マニュアルにあるように,コーシー分布は 2 つのパラメーターを調節して,山の頂点となる最頻値を化石層序の年代よりすぐ古くに設定したり,それよりも古い分布を長い tail として表すことができます.まずは Evolution 2009 のスライド(1.2MB) をご覧下さい.論文で用いた Fish data はこちらからダウンロードしてください.
詳しくは以下のコラムを見てください.
Inoue, J.G., Donoghue, P.C.J., Yang, Z. 2010. The impact of the representation of fossil calibration on Bayesian estimation of species divergence times. Systematic Bioloy, in press.
- (この問題は解決されたようです.[2008 年 5 月])
Approximate likelihood caliculation を codeml で試したところ,まだ不完全なのか解析が説明書通りには動きません.
Sequences read..
Counting site patterns.. 0:00
469 site patterns at 3331 sites, 0:00
Counting frequencies..
Are these a.a. sequences?
469 patterns, clean
AAML in paml 3.14, January 2004
ns = 7 ls = 469
Bad option 'P' in first line of seqfile |
あるいは
Sequences read..
Counting site patterns.. 0:00
469 site patterns at 3331 sites, 0:00
Counting frequencies..
Are these a.a. sequences?
469 patterns, clean
sh: codeml: command not found |
というメッセージが出ましたが,partition ごとの各種 infile は作成されていました.この infile を使って解析を続行することは可能です.ただ,ratematrix に数字が記されており,これを mtREV24.dat などに変更して解析を行いました.
- Approximate likelihood calculation を用いない場合は,解析速度が非常に遅く,ほとんど実用的ではないそうです (論文かマニュアルに書いてありました).10, 000 塩基, 40 OTU のデータを用いて,4000 iterations を burnin した後で 40,000 iterations の解析を行ったところ,5 日ほどかかりました.
|
|
|
- 解析の例題は,「paml4/examples/DatingSoftBound」に基本的なファイル入っていま.各種 infile の作成はこの例題とマニュアル (paml4) を参照して下さい.また,最近になって step-by-step manual を作りました.こちらの下の方にあります (2011 年 7 月).
- ctl ファイル の各種 パラメーターについては paml4 のマニュアルの他にも,README か Rannala and Yang (2007) が参考になります.
- Approximate likelihood calculation のやり方は,以下のコラムをご覧下さい.
- finetune については,次のコラムをご覧下さい.
|
|
|
注意:Version 44e から fineture を自動的に調節するオプションがつきました.mcmctree.trc ファイルにある finetue に,数字とコロンが加わりました.0 は手動で調節,1 は自動調節みたいです.詳しくはマニュアルをご覧下さい.同じファイルを古い version で動かすとおかしな acceptance proportion が得られるので注意してください.自動調節はまだ十分な検討はなされていないようです.
finetune = 0: 0.04 0.2 0.3 0.1 0.3
ctl ファイルにある finetune の設定は重要です.これらの値は MCMC サンプリングに際して,各種パラメーターをどれぐらい次の generation でジャンプさせるかを決めているそうです.
finetune = 0: .02 0.08 0.08 0.1 .4 * times, rates, mixing, paras, RateParas
それぞれ MCMC サンプリングの step length を示すようです.
(a) times
change the species divergence times
(b) rates
change the rates
(c) mixing
performs the mixing step
(d) paras
change substitution parameters (such as k [カッパ] κ and α [アルファ] in HKY+ Γ)
もし JC モデルなどを選択している場合は acceptance proportion は 0 になります.
(e) RateParas
change parameters in the rate-drift model (for clock = 2 or 3 only)
finetune の設定は Exact でも Approximate の解析どちらでも行う必要があります.計算をスタートさせると画面上に以下のような値が出ます.この左から 5 つの値 (acceptance proportion と言うらしいです) がそれぞれ 0.2〜0.4 の間にないと得られる結果が信用できないそうです.
|
|
|
burnin と nsample をそれぞれ 2 桁ぐらい小さくさせて,% で表されている解析の進行状況が 30 秒ぐらいで 50 % ほど出るようにします.ctl ファイルにある finetue の該当の数字 (例: outifle の左から 2 番目の数字が大きすぎる場合は,ctl ファイルでも 2 番目の数字を小さくする).この動作を何度か行って,左から 5 番目までの数字 (下の図では赤で囲ってあります) が それぞれ0.2〜0.4 の間にくるようにします.0.3 である必要はなく 0.39 でも良いみたいです.なお下の例では,4 番目と 5 番目がずっと 0 になっています.これらは解析に使われていないそうで,0 の場合は clt ファイルの finetune を調節する必要はないそうです.
ただ,データセットによってはどうしても acceptance proportion が 0.2〜0.4 の間に来ないことがあります.その場合は acceptance proportion が 0.1〜0.9 ぐらいの範囲に収まっていれば良いとのことです.この場合も,同じ解析を 2 回行い,同様の結果が得られているかどうか確認すれば良いとのことです.ただ,acceptance proportion の 値が変化しなくても,ctrl ファイルに記入する finetune の数字を極端に大きくしたり小さくするのは良くないみたいです.
finetune については, MCMCcoal (Yang さんが作成したプログラム) のマニュアル p10, "Adjusting finetune parameters" に詳しく書いてあるそうです (2008 年 4 月).
=> 最近は MCMCTREE のマニュアルの方が最新の話題をフォローしているようです (2011 年 6 月).
以下の文献も参照してください.
Rannala, B., and Z. H. Yang. 2003. Bayes estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164:1645-1656.
Thorne, J. L., H. Kishino, and I. S. Painter. 1998. Estimating the rate of evolution of the rate of molecular evolution. Molecular Biology and Evolution 15:1647-1657.
|
|
|
- Sequence ファイルは「paml3.15/examples/SoftBound/mtCDNApri123.txt」を参照してください.Phylip 形式と類似していますが,3 partitions (領域)の場合は Sequential の配列を 3 セット作ることになります.
- 1 partition で解析を行う場合は,multidistribute の sequence file がそのまま使えます.アミノ酸配列も同様です.アミノ酸配列の例題はこちらを参照して下さい.
|
|
|
- 化石制約を tree file に newick format の tree と共に書きます.この辺りは version によって変更されることが多いので,ダウンロードして得られるマニュアルをチェックする必要があります (2011 年 7 月).
- 根幹の分岐も含めて,系統樹は完全 2 分岐にしておきます.多分岐が含まれていると作動しないようです (GSF).
- multidistribute の一連の操作で,最後に行う multidivtime の mcmc 解析でも外群を外しました.mcmctree でも同様に外群を外して解析を行います (Manual ver.4 P33).
- 年代の制約は tree ファイルに書き込みます.paml4 のマニュアルで指摘されているように,constraint を「'」でくくっておくと,TreeView で確認することができます.私が解析を行った範囲では,ゼロを省略せずに「>.06」を「>0.06」としても問題なく作動しました.
- コーシー分布はデフォルトで p=0.1, c=1 になっています.もし 0.06 を下限とした制約を作る場合,
L(0.06)
L(0.06, 0.1, 1)
と tree file に書き込みます.
p=0.5, c= 0.2 の場合は,
L(0.06, 0.5, 0.2)
((((human, (chimpanzee, bonobo)) 'L(0.06, 0.1, 1)', gorilla), (orangutan, sumatran)) 'L(0.08, 0.5, 0.2)', gibbon);
詳しくは以下のコラム「Hard/Soft bound 設定」をご覧下さい.
- Upper や Lower constraint をかけた node に,さらに Gamma constraint を制約として施すことはできません (2009 年 5 月).
|
|
|
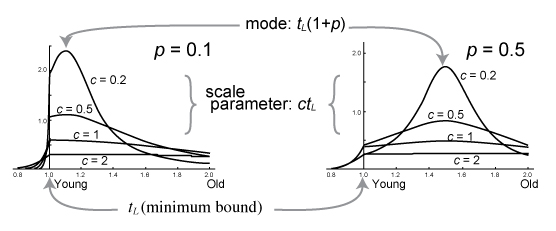
コーシー分布は下限制約に施す分布です.「この化石は信頼できる」,とか,「化石だけから考えると XX Mya ぐらいに分岐がありそう」,などといった化石の情報を年代推定に盛り込むのに分布の形が適しているためコーシー分布が選ばれました. L( tL, p, c) という形でパラメーターを newick format の tree に書き込みます.
密度関数
コーシー分布による下限制約 t > tL の密度関数は以下で表されます (Inoue et al. 2010 の式 26).
分布
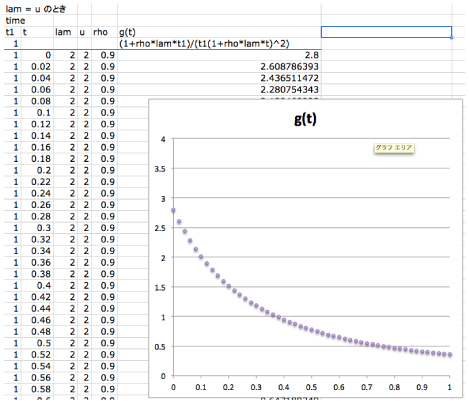
グラフで表すと,以下のような分布になります.Inoue et al. (2010) でパラメータを変化させたところ,c は p よりも prior や posterior に対する影響が大きかったです.
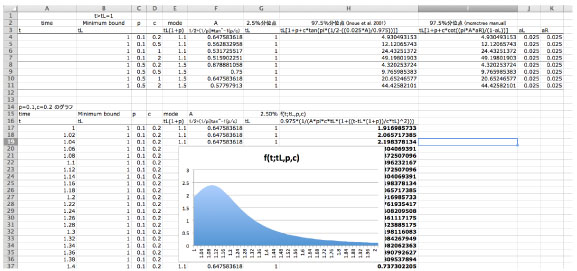
エクセルによる 97.5% 分位点の計算とコーシー分布曲線の作成
以下の図にあるように,上の図の条件 (p=0.1,0.5; c=0.2,0.5,1,2) で 97.5% 分位点をエクセルで計算しました.エクセルファイルはこちらです.論文の値はミスがあるようなので,訂正の記事がそのうち出るそうです [mcmctree の公式マニュアルに数値が出ていました.私の計算と一致していました (2011 年 8 月)].その下には,p=0.1,c=0.2 のグラフを作りました.p や c の値の列をすべて置き換えることで,グラフが作成されます.このグラフをコピー & ペーストで Illustrator などに持ってゆくことができます.
分位点を求める式
|
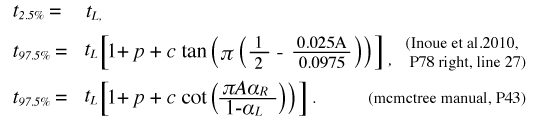 |
|
実際の解析
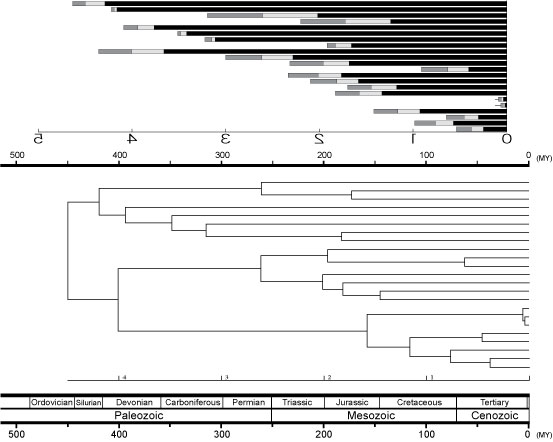
実際の解析で,コーシー分布の事前分布と事後分布でどのように変化するのか見てみました.ここでは Zebrafish/ Stickleback の分岐に 3 種類の分布(左から,Cauchy(ρ= 0.1),Cauchy(ρ= 0.5),Uniform) を当てはめています.上から 3 種類の解析結果が示されていて,それぞれ,
・エクセルで作成された理論的な事前分布 (Specified priors for Zebrafish/Fugu split),
・MCMCTREE usedata=0 解析結果 (データを使わないで得られた事前分布),
・usedata=2 の結果 (データを使った得られた事後分布)
です.prior や posterior の分布は mcmc.out ファイルを Tracer で解析して得られます.用いたデータは実際の核遺伝子データ (Ex. 3rd, ca. 2000bp) を使っています (4spp_folC.tar.gz).このデーターは Unix で解析しています (Mac version では動きませんでした).もしかしたら 4spp のように少ない OTU では Mac version は動かないかも知れません [2011 年 1 月].
Uniform 事前分布を用いた場合,Zebrafish/Fugu 分岐で得られた事後確率は,Cauchy よりもずいぶん広がった分布となっています.よく見ると,Human/ Fugu の分岐で得られた事後分布は,Zebrafish/ Fugu で与えた事前確率によって形が異なっていることがわかります.データに含まれる系統的な情報がどれぐらいあるかにもよりますが,事後分布は事前分布の形に結構左右されると言えるともいます.ちなみに,このデータを用いて系統樹探索を行うと,広く受け入れられている系統関係が統計的に高い値で支持されました.
|

|
|
出生死滅過程 (birth-death process) というモデルを用いて,MCMC 解析のプライアーとなる時間軸付き系統樹を選びます.コントロールファイルの
BDparas = 2 2 .1 * birth, death, sampling
に該当します.BS para = MCMCTREE ではトポロジーは常に固定されているので,プライアーとなるそれぞれの節の分岐年代を BD process によって選んでいます.内部枝の詰まった tree か,あるいは末端の枝が短い tree にするかを決めるパラメータと言えます.
λ: 系統あたりの出生 (種分化) 率 (per-lineage birth (speciation) rate)
μ: 系統あたりの死滅 (絶滅) 率 (per-lineage death (extinction) rate)
ρ: 標本抽出される割合 (sampling)
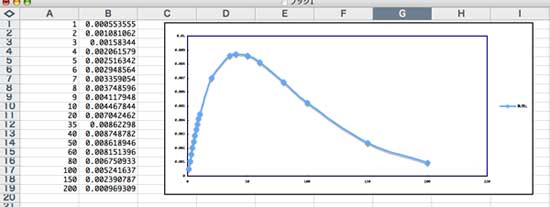
詳細な式は藤ら (2009) P238 あたりを参照してください.ここではλ=μの場合の式 (以下) とカーネル密度のグラフをエクセル (MCMCTREE_BDparas.xlsx)で書いておきます (λ!=μの場合はうまくいきませんでした).
|
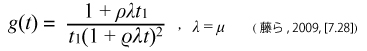 |
このカーネル密度から標本を選んでいるらしいのですが,具体的な部分は私にもよくわからないです.おそらく横軸は分岐年代で,1 が根幹の年代 t1 (ここでは t1=1 としてしている) となっており,それぞれの分岐年代が頻度 (縦軸) にあわせて MCMC 解析でサンプリングされるのだと思います.
ρ=0 で解析を行っても問題ないです.Inoue et al. (2010) では,MULTIDIVTIME で用いられているディリクレ分布 (分岐のテンポを均等にする) に合わせるため,λ= μ= 1,ρ=0 で解析を行っています.「ρ=0 ではまずいのでは」という指摘を以前いただきましたが,上記の式で ρ=0 も問題ないように思えますし,エクセルファイルで ρ の部分を 0 にしても,ディリクレを示すカーネル密度 (フラット) が得られました. |
|
|
|
- コントロールファイルで指定した outfile (outfile=) に必要な情報が入っています.しかし,以下のように Fossil calibration information を得るために,スクリーンアウトも保存する必要があります.
として動作確認をしたあと,command+C で一度解析をストップさせます.そして,
として解析をスタートさせる必要があります.srout にスクリーンアウトが 保存されます.& はバックグラウンドで解析を行うコマンドです.
- 解析が終了したら,Fossil calibration information をチェックした方が良いです.スクリーンアウトに以下のように出力されます.
Fossil calibration information read from the tree.
Node 110: B ( 4.1900, 4.7200)
Node 113: B ( 3.9200, 4.5000)
Node 116: B ( 3.4500, 4.0700)
Node 118: L ( 1.3000, 0.0000)
Node 121: L ( 2.8400, 0.0000)
Node 124: L ( 1.1200, 0.0000)
Node 125: L ( 0.9400, 0.0000)
Node 126: L ( 1.5100, 0.0000) |
node number はコントロールファイルで指定した outfile に書かれています.
|
| mcmc.out の解析 |
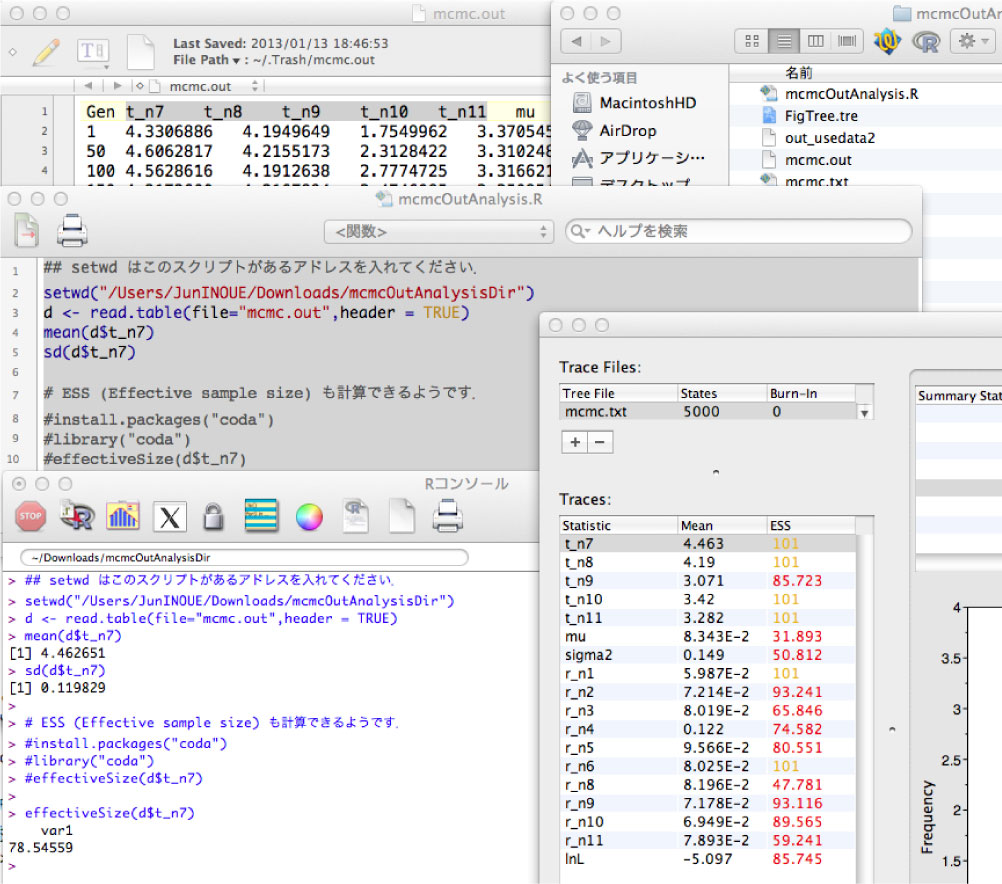
R を使った解析
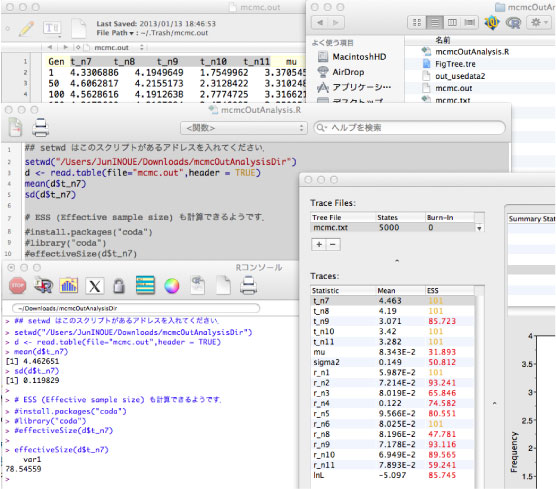
mcmc.out ファイルにはサンプリングされた値が書かれています.tracer で解析するのが楽ですが,サンプル数が多いときは tracer では読み切れないので R で解析します.R のインストールに関してはこちらをご覧ください.
mcmcOutAnalysisDir.tar.gz
Mac の場合は,mcmcOutanalysis.R ファイルを R にドラッグ&ドロップしてください.このとき,setwd("/Users/JunINOUE/Downloads/mcmcOutAnalysisDir") の "〜" 部分を,解析を行っているディレクトリのアドレスに書き換えてください (Unix であれば,pwd で調べることができます).そしてコマンド+A ですべてのスクリプトを選択しリターンを押すと,コンソール (R の出力画面) に答えが出ます (下図を参照).
mcmcOutAnalysis.R ファイルの「d$t_n7」を「d$t_n8」などに書き換えることで,異なる列を解析できます.ここで 「t_n7」や「t_n8」 は最初の行に書いてある列の名前です.
ESS (effective sample size) も coda というパッケージにある effectiveSize で解析できるようですが,tracer と得られる値が異なっていました.今のところどうして違いがでるのかわからないです [2013 年 1 月].
|
|
|
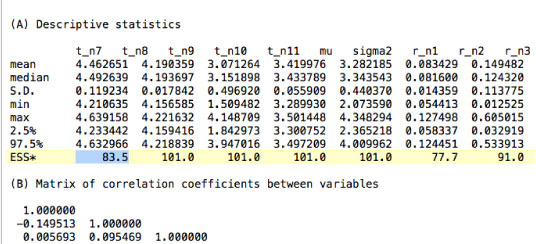
ds を使った解析
paml パッケージの一部である ds というプログラムでも解析することができます.
ds は paml/src に入ってコンパイルを行う必要があります.
cc -o ds -O2 ds.c tools.c -lm
でコンパイルできます.Mountain lion では cc だとプログラムが空のアウトプットを出しました.cc を gcc に変えてコンパイルすると,正常なアウトプットファイルが得られました.
その後,
ds mcmc.out
によって解析が行われ,out.txt に mean や ESS が算出されます (ESS は,Tracer, R, ds の 3 つで異なる値が得られてしまいました).
tracer で解析を行うこともできます.いずれの場合も,Effective Sample Size は 200 以上であれば良いそうです.こちらをご覧下さい. [2013 年 1 月]
|
|
| Approximate likelihood calculation |
|
解析速度はかなり速く,実質はこのオプションを使うことになると思います.塩基とアミノ酸両方の解析が可能です.
paml4 では,multidistribute のように尤度を multidimensional normal distribution (多次元正規分布) を用いて概算するオプション (Approximate likelihood calculation: usedata =2 と 3) が採用されています.Exact calculation (usedata = 1) に比べ数十倍ぐらい速いです.解析の手順も multidistribute と類似していて,解析が二段階からなっています.
(1) 最尤推定値における樹長と分散共分散行列の推定 (usedata =3 解析)
ctl ファイル にある usedata を usedata =3 としして,最尤推定値 (maximum likelihood estimate; MLE) における枝長とその分散共分散行列を計算します.mcmctree を走らせますが,実際には baseml (codeml) が計算を行うため,baseml のインファイルが mcmctree によって作成されます.このため同じフォルダーに baseml を入れておく必要があります.baseml を mcmctree と各種 infile と同じフォルダに入れます.あるいは PATH をきる必要があります.PATH の切り方は,MAFFT のページを参照してください.
(2) MCMC アルゴリズム
usedata = 2 としてベイズ解析を走らせて年代推定を行います.解析をスタートされる前に (1) で出力された out.BV を in.BV という名前に変更してください.
|
|
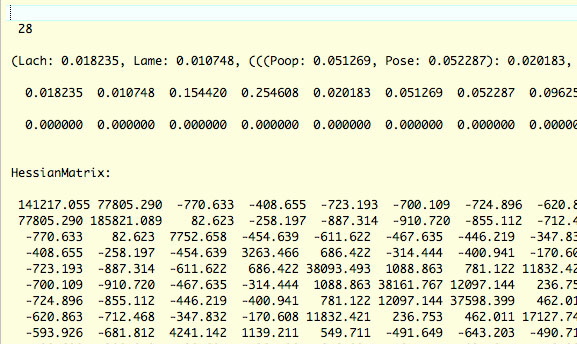
out.BV ファイルについて
usedata =3 解析で得られた結果をようやくしたファイルです.これが usedata =2 の解析に用いられます.種数,枝長付き tree,枝長,枝長の傾き,Hessian matrix が書かれています.
枝長
枝長は単に枝長付き系統樹から取り出してきた数値です.0 になっていると,良くないそうです.マイナスもダメです.
3 行目の枝長は,単に 2 行目の数字だけを書き直しているだけです.同じ数字があります.
枝長の傾き (gradient)
枝長の gradient は 0 になっている必要があります (下図,4 行目).これは,例えば,x 軸に枝長,y 軸に尤度をとって,尤度を最適化した場合,曲線の gradient を示すそうです.gradient が 0 である場合は,尤度が最適化されたことを意味するそうです.例えば,尤度推定の際に枝長を固定してしまうと,この値が 0 にならないことがあります.しかし,すべてが 0 でなくても,小さな数字であれば良い,と Yang さんはおっしゃっていたように思いますが,私は確かめたことがありません.
ゲノムデータのようなとても長いデータを扱った場合 (100 万 bp 以上でしょうか),尤度を表す山が非常に尖ったものになります.そうすると通常の設定ではHesian Matrix の桁数と gradient の桁数がうまくあわず,baseml の計算が最尤値にたどり着かなくなることがあります.この場合はアウトファイルに「Convergence?」と出てしまいます.現在この問題を解決しようと Yang さんがプログラムを変更なさっている最中です (2010 年 2 月).
Hessian Matrix
枝長の分散・共分散マトリクスです.求め方は傾きと一緒で,枝長を少しだけ変化させていって,分散・共分散を求めるようです.該当する文献をまだ見つけていないので,詳しくは調べる必要があります.
以下のように gradient が 0 でないこともあるようです.これぐらいの数字であれば,MCMC 解析 (usedata = 2) の in.BV ファイルとして使ってよいということでした.
|
|
|
自動的に作られる tmp*.* ファイルについて
* tmp.tree はオリジナルの tree ファイルを unrooted として保存しています.これに関しては Computational Molecular Evolution の第七章に詳しい説明があります.
*tmp.txt はサイトパターンがまとめられています.このため,サイト数はオリジナルの配列データより少なくなっていますが,ファイルの末尾に,サイトパターン数と同じ数字列が記載されています.これはそれぞれのサイトが何回オリジナルのアライメントで出現したかを示しています.
|
|
|
mcmctree の化石制約は Hard と Soft を選ぶことができます.2009 年ぐらいまではコンパイル方法によって Hard か Soft を選んでいましたが,現在は tree ファイルにコマンド書き込むことで Hard か Soft を選択できます.私の知る限り,コーシー分布を化石の事前分布として用いた場合しか適用できないみたいです.詳しくはマニュアルの P43 をご覧下さい.デフォルトでは soft bound (tail probatility 0.025) です.
下限制約 (Minimum boundary)
L(tL, p, c, pL)
記述方法から説明します.これは Minimum-age bound,tL を決定します.
p: コーシー分布の location parameter tL(1 + p) を決めるパラメーター
c: コーシー分布の scale parameter ctL を決定します.
pL: Left tail probability.
Soft Bound: L(0.06, 0.1, 0.5, 0.025)
Hard Bound:
L(0.06, 0.1, 0.5, 1e-300)
Soft bound の tail probability (グラフを書いたときに左になるので,left tail probability と言うみたいです) の 0.025 はデフォルトなので,L(0.06, 0.1, 0.5) と同じ意味です.
Hard の 1e-300 はとても小さな数字を表しています.0 ではいけません.0.1 などの tail probability を選ぶこともできるようですが,私は試したことがありません.
上限制約 (Maximum boundary)
U(tU, pR) は maximum-age bound, tU を決定します. pR は right tail probability です.
U(0.08, 0.025)
私は試したことがありませんが,U(0.08, 0.025, 1e-300) で Hard bound になると思います.
ペア制約 (Paired boundary)
B(tL, tU, pL, pU) で pair bound を表します.tL と tU は maximum と minimum bound です. pL と pU はそれぞれの tail probabilities です.
B(0.06, 0.08, 0.025, 0.025)
これも 1e-300 を用いることで,Hard bound になったと思います.
(2011 年 7 月)
|
|
|
(ガンマ分布の記述は,少し偏った内容になっています.しかも,ここにあるような方法で分布の形を調節することは少なくなっていると思うので,初めての人はわからない部分は飛ばして読んでください)
以下では,ガンマ分布を仮定して,制約として用いる年代の 2.5% 点 (97.5% 点) を計算する方法を紹介しています.化石記録から制約をかけられないけれど,root node の年代の事前分布を狭くしたい場合に役に立つと思います (実際にはあまり用いている例は見かけません).
mcmctree はデリクレを default の root node age の事前分布にしているので,root node に制約がないと事後分布として得られる root node age の信頼区間が multidivtime (事前分布にガンマ分布を使っている) に比べて広くなってしまいます.なお個人的な意見ですが,曲線の形が事後確率を狭くするのに都合良いという以外に,ガンマ分布を選ぶはっきりとした根拠はないように思います.
もっともあり得そうな年代を平均値として,lower (2.5%) と upper (97.5) bound の年代を R の qgamma 関数を使って計算します.qgamma はガンマ分布の累積分布関数 (CDF: cumulative density function) の分位関数 (quantile function) (逆関数のこと) で,信頼区間の確率 (%),shape (α),scale (β),から,lower (あるいは upper) とする年代を求めることができます (R の確率分布にかんしてはこちらをご覧下さい).統計解析システム R では qgamma の各種パラメーターは,
qgamma(p, shape, scale, lower.tail, log.p)
となっています.p=2.5 (%), m=42, s = 42 の場合は,shape parameter のαと scale parameter のβは以下のようになります.
α=(m/s)^2
=(42/42)^2
=1
β=m/s^2
=42/42^2
=1/42
これらの値を R のコマンドに加えます.
> qgamma(0.025,1,1/42,lower.tail=TRUE,log.p=FALSE)
[1] 1.063348
となりました.
こうして得られた値を,tree file 内部に制約として書き込みます.mode (mean としていましたが,プログラムはこの値を mode [peak のこと] として扱います) は 42 にしたいところですが,少し小さい値を選んで 41 ぐらいにするように,と言われました.理由はよくわかりません.
'>1.063348=41<154.9329'
計算が終了したら,screen out に mcmctree で用いられたガンマ分布のパラメーターが出力されます (この例ではおかしな値になっています.いまプログラムを 修正しているところだそうです [2008 年 7 月]).
Fitting gamma to '>1.0633=42.0000<154.9329'
a = 2.501893 b = 0.035759 CDFs (0.000082 0.950000 0.950082)
なお,より簡単に Upper と lower を Excel から計算することも可能です.
挿入 -> 関数... から統計 -> GAMMAINV を選びます
確率に 0.025, A に 1, B に 42 を入れると R と同様の値が得られます.ここで注意したいのが,scale parameter である βがエクセルでは 1/βとして扱われている点です.これは人によるようで,shape parameter と scale parameter をα,βとすることもあれば,α, 1/β とする場合もあるようです.統計プログラムによって思い通りの数字が得られない場合は,βを 1/β として計算させ直す必要があるみたいです.
余談ですが,図のように適当なプロットをつくって,ガンマ分布を Excel で作成することが出来ます.B 列は =GAMMADIST(A1,2,42,0) という関数になっています.
|
|
|
Inoue et al. (2010) では,Prior estimates を表示することが重要であるとされています.ここで言う Prior estimates とは,データを用いないでベイズ推定した値のことです.Prior analysis で得られた time tree と 95% 信頼区間を見ることによって,化石情報の設定が正しいかどうか見ることができます.
Prior estimates を得るには,コントロールファイルにある以下パラメータを設定し直してください.
usedata = 0 * 0: no data; 1:seq like; 2:normal approximation
in.BV ファイルが無くても解析が始まります.
sampfreq = 20
posterior 解析の 10 倍必要です.20 である必要はありませんが,もし posterior を sampfreq = 2 で推定するなら,sampfreq = 20 にしてください.
|
| 大規模データの解析: Approximate 法のパラメーター推定 |
|
Approximate 法を用いても解析が終わらない場合は,baseml や codeml を用いた第一段階の解析 (パラメーター推定: usedata = 3) を工夫する必要があります.二段階目の解析 (usedata = 2) は,データの大きさにそれほど左右されないので,工夫する必要はないようです.
パーティションごとに手動で baseml (codeml) の解析を行う
mcmctree を走らせた後で一度解析をストップし,独自に baseml や codeml を model =1 で走らせて Hessian matrix (通常の Approximate 解析では out.BV として出力される) を得ます.ここでは 4 パーティションで解析した場合を例に説明します.
1.
Approximate で mcmctree を走らせ,tmp1.ctl などのファイルができたら解析をとめます.
2.
tmp1.ctl の method を method = 1 にして,
baseml tmp.ctl
あるいは
codeml tmp.ctl
などとして,baseml/ codeml を mcmctree とは独立して走らせます.
3.
rst2 が作成されたら,これを in.BV という名前に変えます.
4.
同様の操作を tmp2.* ファイルを用いて行います.得られた rst2 の内容を,in.BV の最後に付け足します.これを他 2 つのパーティションについても行って,in.BV ファイルを完成させます.
Tree file について
mcmctree を usedata = 3 で走らせると,根幹が 3 分岐となった unrooted tree が tmp.trees に書き換えられて,これが baseml や codeml の解析に用いられます.通常 mcmctree が自動に行ってくれる baseml や codeml の解析を行うには,RAxML などで得られた tree が unrooted になっているか確認する必要があります.
[2010 年 1 月]
さらに少しだけ解析を速くする
上記の手順で解析をするのですが,codeml (baseml) を二段階に分けて解析します.一段階目で one rate model (サイト間の進化速度差異を補正しない) で枝長を概算します.この枝長付き tree を starting point として,ガンマモデルを用いた二段階目の解析を行います.4 パーティションであれば,各パーティションごとに以下の手順を行うので,少し面倒です.
1.
tmp.ctl を以下のように書き換えて,最初の解析を行ってください.
seqfile = ../tmp1.aa.txt
treefile = ../tmp1.trees
outfile = out
noisy = 3
seqtype = 2
model = 3
aaRatefile = ../wag.dat
fix_alpha = 1
alpha = 0
ncatG = 5
Small_Diff = 0.1e-6
getSE = 0
method = 1
2.
最初の解析が終了したら,output file から枝長付きの newick format tree をコピーして,tmp.tree にある枝長なしの tree と交換します.そして tmp.ctl を以下のように書き換えて,解析を再度スタートします.
seqfile = ../tmp1.aa.txt
treefile = ../tmp1.trees
outfile = out
noisy = 3
seqtype = 2
model = 3
aaRatefile = ../wag.dat
fix_alpha = 0
alpha = 0.5
ncatG = 5
Small_Diff = 0.1e-6
getSE = 2
method = 0
rst2 が得られます.これを in.BV という名前にして,usedata = 2 の解析に用います.
RAxML などで推定した枝長を利用して Hessian matrix を得る
RAxML など解析速度の速いプログラムで推定した枝長付き系統樹を利用して,各種パラメーターを推定します.解析速度は速くなりますが,パラメーターの尤度推定は概算になってしまいます. こちらの例題にそって説明します (in.codeml というファイルは in1.codeml という名前に変更してあるので,このファイルを 3 の解析に用いる場合は in.codeml という名前に変更してください).
RAxML で GTRGAMMA の解析を行うと,GTR + G4 (4 カテゴリー) のモデルになります.baseml の解析で,進化モデルを代わりに F84 などを選んでもそれほど問題になることはないそうですが,ガンマのカテゴリー数はあわせておく必要があるそうです.
準備.
codeml.c (あるいは baseml.c) を書き換えて,枝長を数字として out.codeml (out.baseml) というファイルに出力できるようにします. コンパイルの後に,codeml (baseml) を「outcodeml (outbaseml)」という名前に書き換えます.
codeml.c の「GetInitials(x, &i);」の下に 「/*」と「*/」にはさまれた行を貼付けます.「/*」と「*/」は,これに挟まれた行はコンパイル時に無視される記号です [2010 年 7 月].
GetInitials(x, &i);
/*
{
FILE *fblength=gfopen("out.codeml", "w");
if(com.fix_blength) fprintf(fblength, "-1\n");
matout(fblength, x, 1, com.np);
exit(0);
}
*/
np = com.np;
if(noisy>=3 && np<100) matout(F0,x,1,np);
baseml.c は以下です.
GetInitials(x, &i);
/*
{
FILE *fblength=gfopen("out.baseml", "w");
if(com.fix_blength) fprintf(fblength, "-1\n");
matout(fblength, x, 1, com.np);
exit(0);
}
*/
if(i==-1) iteration=0;
if((np=com.np)>NP) error2("raise NP and recompile");
枝長付き系統樹を tree ファイルに保存します.unrooted tree (根幹三分岐) であることを確認してください.rooted tree だと rst2 に Hessian matrix が出てきません.例題にある stweart.BLtree を参照してください.
1.
枝長を固定して,サイト間進化速度を補正するのに用いるガンマのパラメータαを推定します.
α (とκ) を推定するだけなので,解析はそれほど時間がかからないと思います.たとえば,baseml を使って 500,000 bp の配列を解析しても,それなりの PC であれば一晩もかからないと思います.codeml は baseml よりも時間がかかります.baseml の outfile に「check convergence..」と出ることもありますが,これはかなり conservative な感じで判断しているので,気にしなくてよいそうです.
コントロールファイルの該当箇所を以下のように変更します.例題 codeml1.ctl を参照してください.
seqfile = stewart.aa
treefile = stewart.BLtree
outfile = out_codeml1
noisy = 3
seqtype = 2
model = 3
aaRatefile = wag.dat
fix_alpha = 0 *[アルファを最尤推定する]
alpha = 0.5 *[0.5 を初期値として最尤推定する]
ncatG = 4
Small_Diff = 0.1e-6
getSE = 0
fix_blength = 2 * 0: ignore, -1: random, 1: initial, 2: fixed
*[stewart.BLtree の枝長を固定する]
method = 1
mlc1 の最後に alpha の値が書かれています.
以下のコマンドを実行して下さい.
./codeml codeml1.ctl
2.
ソースコードを書き換えて得られた outcodeml を用いて,枝長を out.codeml に書き出します.RAxML の枝長などは,四捨五入した値になっています.書き換えるだけなので,瞬間的に解析が終わるはずです.
コントロールファイルの該当箇所を以下のように変更します.例題 codeml2.ctl を参照してください.
seqfile = stewart.aa
treefile = stewart.BLtree
outfile = out_codeml2
noisy = 3
seqtype = 2
model = 3
aaRatefile = wag.dat
fix_alpha = 1*[αを固定して,計算負荷を減らします]
alpha = 0.5
ncatG = 4
Small_Diff = 0.1e-6
getSE = 0
fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed
*[2 が枝長固定ですが,2 だと 枝長が out.codeml に書き出されません.outcodeml では fix_blenght = 1 でも枝長が推定されず,枝長の初期値がそのまま out.codeml に書き出されます]
method = 1
以下のコマンドを実行して下さい.
得られた out.codeml を in.codeml という名前に変更します.in.codeml の一行目 (-1 と書かれている) は削除してください.in.codeml の一行目が -1 となっていると,in.codeml を下の 3 の解析に用いる場合,ここに書かれている各種パラメーターを fix して解析を行うことになってしまいます.詳しくはマニュアル「Specifying initial values」をご覧下さい. in.codeml については,マニュアルにも書いてあります (in.codeml を使わない解析を行う場合は,名前を変えておく必要があります).
補足:
out.codeml の内容は,通常のコンパイルで得られた codeml を用いて作成することもできます.通常の codeml を走らせると,スクリーンアウトの「Reading matrix from wag.dat」の下の行に枝長の数字のみが書き出されます.例題にある out.codeml も参照してください.しかし誤りが多いと思うので,なるべく上記の outcodeml というプログラムを用いて解析した方が無難です.
0.009296 0.022476 0.081276 0.033827 0.063295 0.276238 0.096219 0.242936 0.592165 0.500000
3.
すべてのパラメーターを固定して,枝長を最適化させてながら (RAxML で推定した枝長を starting point に用います) Hessian matrix を推定します.1 で得られた α の値 (baseml であれば κ の値も) (これらは手入力してください) と,2 で得られた枝長が記入された in.codeml を用います.Hessian matrix の推定は,若干時間がかかります.
コントロールファイルの該当箇所を以下のように変更します.例題 codeml3.ctl を参照してください.
seqfile = stewart.aa
treefile = stewart.BLtree
outfile = out_codeml3
noisy = 3
seqtype = 2
model = 3
aaRatefile = wag.dat
*fix_kappa = 1 * 1: kappa fixed, 0: kappa to be estimated
*kappa = 11.00818 * initial or fixed kappa
fix_alpha = 1 *[αを固定します]
alpha = 1.64704 *[1 の手順で推定されたαの値を書きます]
ncatG = 4
Small_Diff = 1e-9
getSE = 2
fix_blength = 1 * 0: ignore, -1: random, 1: initial, 2: fixed
*[2 が枝長固定ですが,2 だと Hessian が出力されません.このため枝長を in.codeml に保存して (1 行目に -1 を書かない) 解析に用いています]
method = 1 *[kappa と alpha を固定して枝長に関するパラメーターだけを推定する場合は,method = 1 の方が 0 よりも良いそうです.]
以下のコマンドを実行して下さい.
./codeml codeml3.ctl
rst2 に Hessian matrix が出力されます.
rst2 の名前を in.BV に変更して,Approximate 法・二段階目の解析に用います.
メモ:
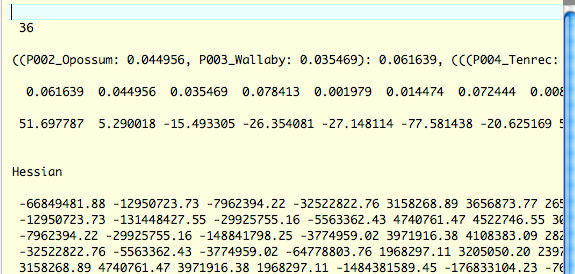
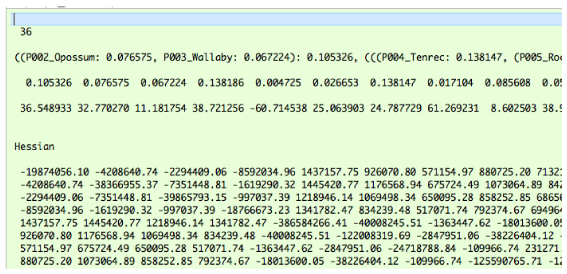
36 種 11,524,095aa のデータでは 3 の解析が遅くて枝長を再推定することはあきらめました.このため,fix_blength =1 で解析を行いますが,in.codeml の 1 行名に -1 を付けて,RAxML で推定した枝長を固定して Hessian を出しました.これを usedata = 2 の解析に用いました.この場合,method = 0 で解析するそうです.この方法で解析を行った場合,以下のような rst2 ファイルが作成されました.これを in.BV と名前を変えて usedata = 2 の解析に用いています.gradient を示す 4 行目 (「36.548933」で始まる行) が 0 になっていないことに注目してください.データが巨大すぎるので,仕方なくこれで解析することにしています.
わかりにくいパラメーター設定について
fix_alpha と alpha
fix_alpha は alpha の値を固定するか推定するか決めます.マニュアルでもっともわかりにくい部分の一つだと思います.
・fix_alpha = 0 の場合
alpha を推定します.
「alpha = 0 以上の整数」の場合は,ncatG で指定したカテゴリー数の discrete gamma として,alpha で与えた数を初期値として alpha を計算します.
「alpha = 0」だとどうなるか確かめたことはありません.おそらく,discrete gamma model を仮定せず,サイト間の進化速度差異を無視して,全てのサイトを同じ rate として解析を行います.そして,alpha の初期値がランダムに選ばれると思います.
・fix_alpha = 1 の場合
alpha を固定します.
「alpha = 0 以上の整数」の場合は,discrete gamma model を仮定することになり,進化速度に基づいて ncatG の数だけカテゴリーを作成してサイトを分類します.
「alph =0」 にすると,discrete gamma model を仮定せず,サイト間の進化速度差異を無視して,全てのサイトを同じ rate として解析を行います.
method = 0 と 1
method =0 と 1 の違いについては,「10 Miscelaneous notes,Analysing large data sets and iteration algorithms」に詳しいです.概して,method = 0 は各枝長さを同時に最尤推定するのに対し,method =1 は枝ごとに最尤推定するようです.このため,パラメータを fix している場合は,model =1 の方が解析速度がずっと速いそうです.理論的には method = 0 と 1 で得られる値は同じだそうです.実際,method = 0 で 57 秒かかった解析が,method =1 では 7 秒で終わりましたが,得られた値はほとんど同じでした.
|
|
(PAML45 から,MCMC アルゴリズムによって time tree が推定された後,FigTree.tre ファイルが作成されるようになれました.これによって,信頼区間付きの time tree が描けるようになりました.もはや私の作成した perl script を使う必要はありません [2012 年 1 月].)
方法1
FigTree を使います.mcmctree のアウトプットから信頼区間入りの newick tree を読み込んで,FigTree に入力可能な Nexus file を作ります.
perl script を作りました.スクリーンアウト (あるいは newick tree だけのファイル) から信頼区間入り newick tree を読み込んで,Nexus format を作るスクリプトです.
perl MctToFigTre.pl screenout_usedata2 > out_scout_usd2
あるいは,「> out_scout_usd2」なしで得られたスクリーンアウト (Nexus format) をコピーして,FigTree の画面にペーストしても良いです.
MctToFigTre_fol.tar.gz
|
方法2
TreeView と Illustrator を使って手作業で作ることもできます.
Tree は TreeView からコピー & ペーストで Illustart に貼付けます.グループ化解除などを使って,適度にばらします.
信頼区間は,テキストファイルで得られた結果を Excel で加工して得ます.Illustrator の Stacked Braph Bar Tool を使って,横の棒グラフを作成しています.

上記 2 つの素材は time scale 付きで得られるので,手作業で scale を合わせています.
|
| mcmctree と multidivtime の推定値 |
|
まったく同じ条件を与えた場合,mcmctree はmultidivtime より古い値を返す傾向があると思います.
MCMCTREE は MULTIDIVTIME とほぼ同じ方法で年代推定が行われますが,異なる点があります.MULTIDIVTIME が 1.根幹の年代を推定し,2. この年代を基準にMCMC 解析を行い,3. 制約に合わない generation を捨てる,というステップを踏んでいるのに対し,MCMCTREE は 1〜3 を MCMC 解析で同時に行っています (mcmctree では,3. generation で用いられるパラメータは,事前分布に合わせてサンプリングされる).
開発当初,Yang さんは,これで MCMCTREE でも MULTIDIVTIME と同じような値が出ると予想していたみたいですが,テストデータを解析してみると実際は MCMCTREE では結構古い値が出てしまいました.この違いを補正するために,MCMCTREE では下限制約などに施す偏った分布を利用して,得られる推定値を若くして MULTIDIVTIME に近づける,という方法をとりました.このため,たとえば下限制約の分布を MULTIDIVTIME と同じフラットにすると,MCMCTREE では古い年代が得られます (下限制約のデフォルトはフラットではありません).化石制約が少ない場合,MCMCTREE はより古い年代を出すはずです (制約が少ないと,分岐間の速度を independent rates か correlated ratesにするか [clock]:,などのパラメータが大きく影響します).
上記は開発をお手伝いした私自身の意見です.勘違いしている部分もあるかも知れませんが,MCMCTREE の方が古い年代が出ることが多いと思います.どちらが正しいかわからないです.解析方法としては MCMCTREE のが洗練されていると思いますが,ただでさえ分子推定は古いと言われるので,正直なところ微妙な気分でもあります.
詳しくは Inoue et al. (2010) を読んでください (2012 年 8 月).
|
|
「sh: baseml: command not found」と表示される
mcmctree が baseml を見つけられない,という意味です..bashrc (あるいは .bash_profile) で,baseml があるディレクトリへ PATH が設定されていません.こちらを参照にして,PATH の設定をしてください.
[i2:newAnalysis]$ ./mcmctree
MCMCTREE in paml version 4.7b, October 2013
....
*** Locus 1 ***
running baseml tmp1.ctl
sh: baseml: command not found
「Error: steplength = 0 in UpdateTimes.」と表示される
以下のスクリーンアウトが得られてしまった場合です.MCMC 解析が始まった後にエラーが出ているので,finetune の設定がおかしいようです.mcmctree.ctl ファイルの finetune を 0 から 1 にしたら,解析が動きました:「 finetune = 1: 0.06 0.5 0.008 0.12 0.4」.
[i2:newAnalysis]$ ./mcmctree
MCMCTREE in paml version 4.7b, October 2013
Reading options from mcmctree.ctl..
Reading master tree.
....
Starting MCMC (np = 14) . . .
finetune steps (time rate mixing para RatePara ...): 0.0000 0.0000 0.0000 0.0000 0.0000
paras: 12 times, 1 mu, 1 sigma2 (& rates, kappa, alpha)
Error: steplength = 0 in UpdateTimes.
|
|
|
|
Inoue, J.G., Donoghue, P.C.J., Yang, Z. 2010. The impact of the representation of fossil calibration on Bayesian estimation of species divergence times. Systematic Bioloy, 59, 74-89. [doi:10.1093/sysbio/syp078 ] [Data]
(Cauchy distribution の導入と年代制約の重要さについて書かれています)
Rannala, B., and Z. Yang. 2007. Inferring Speciation Times under an Episodic Molecular Clock. Syst. Biol. 56: 453-466.
(control file のパラメータ設定が説明されています)
Yang, Z. H. and A. D. Yoder (2003). Comparison of likelihood and Bayesian methods for estimating divergence times using multiple gene loci and calibration points, with application to a radiation of cute-looking mouse lemur species. Systematic Biology 52(5): 705–716.
multidivtime の解析ステップとその解説が詳しく書かれています.MCMCTREE の理解にも役立ちます.
Yang, Z., and B. Rannala. 2006. Bayesian estimation of species divergence times under a molecular clock using multiple fossil calibrations with soft bounds. Mol. Biol. Evol. 23: 212-226.
Zhong, B., T. Yonezawa, Y. Zhong, and M. Hasegawa. 2009. Episodic evolution and adaptation of chloroplast genomes in ancestral grasses. PLoS ONE 4:e5297.
|
|
|
|