seed = -1
seqfile = Johnson.txt
treefile = Johnson.tre
outfile = out
ndata = 1
パーティションの数を設定します.
usedata = 1 * 0: no data; 1:seq like; 2:normal approximation
Normal apprixomation の解析 を行う場合は,まず usedata =3 でパラメータ推定の解析を行い (解析自体は baseml か codeml が自動的に行います),次に usedata =2 の解析を行います.Exact (Approximate ではない) 解析は usedata = 1 です.詳しくはこちらをご覧ください.
Prior の設定がどのようになっているか知りたければ,usedata = 0 で解析します.この場合,データを用いないで Timetree が Prior として算出されるようです.sampfreq を 10 倍にする必要があります.この場合も計算が進行中に出る finetune の値が 0.2〜 0.4の間に来るようにします.詳しくはこちらをご覧下さい.
clock = 2 * 1: global clock; 2: independent rates; 3: correlated rates
枝ごとの進化速度を設定します
RootAge = '<1.0' * constraint on root age, used if no fossil for root.
Root node が取りうる最大の年代です.multidivtime では "bigtime" にあたります.Root node に upper bound を制約としてかけている場合は,この upper bound より大きな値であれば,どのような値でも結果に影響を与えないようです.upper constraint よりもゆるい制約といえます.
model = 4 * 0:JC69, 1:K80, 2:F81, 3:F84, 4:HKY85
モデルを選択します.mcmctree マニュアルにはまだ書いていないようですが,塩基配列の解析で GTR モデルを選ぶこともできます.model = 7 です.マニュアル baseml の箇所を参照してください [2010 年 9 月].
alpha = 0.5 * alpha for gamma rates at sites
サイト間の進化速度差異を行うガンマ補正に用いられる shape パラメーター α の値を入れます.この値は,おそらく baseml などを用いてあらかじめ推定したおhだと思います (あまりよく覚えていません).
Approximate likelihood calculation では,usedata=3 解析でこの値を初期値としてアルファの値が最尤推定されます.パーティションに分けた場合は,パーティションごとにアルファの値は推定されるようです.tmp1.out などの最後に,パーティションごとに異なったアルファの値が出ています.ただ,Approximate likelihood calculation の場合,アルファの情報はすべて樹長およびその分散・共分散マトリクスに集約されて MCMC アルゴリズム計算がなされます. Exact calculation (usedata=1) の場合は,MCMC アルゴリズムを用いてアルファの値も最適化されるようです.詳しくは doc/MCMCtreeDOC.pdf P45 「alpha_gamma 」をご覧下さい.(2012 年 8 月).
ncatG = 5 * No. categories in discrete gamma
サイト間進化速度補正のカテゴリー数を入れます.
cleandata = 0 * remove sites with ambiguity data (1:yes, 0:no)?
? や -, N など ambiguous な形質をすべて解析から除外するかどうか設定します.核遺伝子では遺伝子自体が missing であるなど,ambiguous な形質がデータ行列に多く含まれる傾向があるので,これは 0 として解析した方が良いと思います.1 で解析したら,解析に用いるサイトが 0 になってしまうことがありました.
mode, alpha, ncatG, cleandata は,usedata 1 と 3 の時は使われますが,usedata 2 では使われません.
BDparas = 2 2 .1 * birth, death, sampling
[1 1 0 で multidivtime とほぼ同じ状態になるそうです]
kappa_gamma = 6 2 * gamma prior for kappa
alpha_gamma = 1 1 * gamma prior for alpha
kappa_gamma と alpha_gamma は,usedata 1 と 3 の時は使われますが,usedata 2 では使われません.
rgene_gamma = 2 2 * gamma prior for rate for genes
Overall (系統樹全体の ?) 速度パラメーター μ を選定するのに使われているガンマプライアーの shape と scale パラメーター (αとβ) を指定します.これは multidivtime の rtrate の設定にあたります.
multidivtime はガンマ分布を平均 (mean) と標準偏差 (standard deviation) で表しているのに対し,mcmctree は G(α, β) の α と β のパラメータで表しています.G (α, β) の α と β は mean (m) と standard deviation (s) から計算することができます.s は multidistribute のマニュアルに従って,m=s とここではしています.
ガンマ分布 G (α, β) の m と s は,
m = α /β
s = √α /β
と表すことができます
(ガンマ分布の性質のようです.良いサイトが見つからなかったので,DeGroot and Schervish [2003] p297 を参照しました).
ここから α と β を m と s で表してみると以下のようになります.
α = (m/s)²
β = m/s²
計算の例題を作りました.
PAML のマニュアル,"mcmctree -> Description of the variables -> finetune " も参考になります.(2008 年 5 月)
sigma2_gamma = 1 10 * gamma prior for sigma^2 (for clock=2 or 3)
パラメーター σ^2 を選定するのに使われているガンマ事前分布の shape と scale パラメーター (αとβ) を指定します.σ^2 によって枝ごとの進化速度がどの程度変異するのかを設定します.σ^2 が大きければ進化速度の変異が大きいことを示します.σ^2 が小さい場合は,枝ごとに見られる進化速度の差異は小さく,ほぼ同じ分子時計を仮定できることを示します.
clock =1 (global clock) を選択した場合は,このプライアーは解析に用いられません.
sigma2_gamma の設定は multidivtime の browmean (nu) の設定にあたります (correlated-rates model [clock]=3 を用いた場合は,まったく同じパラメーターです).私は multidivtime のマニュアルを参考に,brownmean (nu) time を計算しています (他に良い方法があるかも知れません.一応の目安に過ぎないと思います).multidivtime のマニュアルでは rttm*brownmean = 1 とされているので,rttm (root node time) から brownmean を計算します.ここでも m=s としています.上記の m と s からαとβを算出する式を参考にして,m と s から G (α, β) の α と β を得ます.
MULTIDIVTIME のマニュアル (multidivtime.readme.pdf) にある該当部分です.
brownmean and brownsd set the mean and standard deviation of the prior distribution for "nu" (the variance in logarithm of rate of molecular evolution for an amount of time t is "nu" multiplied by t). We are still working on what is a good choice for the value of brownmean. My current favorite strategy is to have rttm*brownmean be about 1 or 2. I wish I had a more reasoned approach. My current approach for setting brownsd is to have it equal to brownmean.
independent-rates model (clock =2) を選択している場合,それぞれの分岐の進化速度は,対数正規分布 log-normal distribution から得られる独立変数です.
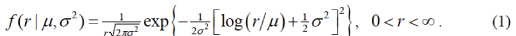
ここで σ^2 は進化速度 r の対数の分散です.
Inoue et al. (2010) Talbe 3 の Flog data set では μ=0.068, σ^2 = 0.24 というプライアーを用いています.
finetune = .2 0.5 0.5 0.1 .9 * times, rates, mixing, paras, RateParas
詳しくはこちらをご覧ください.
print = 1
burnin = 50000
sampfreq = 2
nsample = 200000
ここでは,最初の 50000 iteration を捨てます.その後,2*200000 iteration を行い,2 回ごとにサンプリングが行われてアウトファイルに出力されます.合計 200000 サンプルが読み込まれ,サマライズされます.マニュアルによると,少なくとも 2000 サンプルは必要ということです (2008 年).
mcmc.out ファイルは巨大になります.このため毎回解析が終わるごとに捨てた方が無難です.あまりに巨大すぎる場合は,sampfreq を 20 などにして,20 iteration ごとにサンプリングさせるようにします.
サンプルの回数を単に増やしたい場合は,nsample を大きくします (2010 年 2 月).
Approximate likelihood calculation (ALC 解析) を行う場合,最初の段階である usedata = 3 の解析で mcmc sampling の設定は関係ないです.
[最適な burnin と sampling の見つけ方は,私も良くわかりません.どこかに,seed number を代えて 2 回解析を行い,同じような値が得られればよい,と書いてあった (聞いた) 気がします.あるいは ds という PAML に含まれているパッケージを用いて mcmc.out を解析し,ESS に達しているかどうかを見る方法もあります.こちらをご覧下さい.
参考までに,39 OTU 19984 塩基のデータでは,以下の設定でとくに Yang さんに変更を進められることはありませんでした .
print = 1
burnin = 50000
sampfreq = 2
nsample = 200000
(2008 年).
ちなみにゲノムデータ (23 Mbp) の解析で,
print = 1, burnin = 100000, sampfreq = 10, nsample = 200000
を試したら,これは長過ぎと指摘されました (2010 年 2 月).
|