|
Supercomputing with SLURM
|
|
|
|
This website explains how to use SuperComputer, DEIGO, in OIST. You need OIST ID and password to see some links in this website.
Your analysis is recommended to conduct under /work directory. The compiled programes can be save under home directory (~).
Some documentations for SANGO can be found from SCS top>Getting started. The Sango cluster runs via SLURM scheduler (Documentation).
Documentation page explains the deigo usage.
|
|
In OIST servers, 50GB is the limit of each user's home. You can find the size of each directory/file at your home by the following command:
$ du -sh ~/*
Also, we can check the size of a directory:
$ du -sh jun-inoue
So we need to check file and delete unnecessary files.
[cluster:~]$ hostname
deigo-login4.oist.jp
[cluster:~]$ du -sh ~/*
16M /home/j/jun-inoue/000geneTree3u
850K /home/j/jun-inoue/100_2ndTree.zip
11G /home/j/jun-inoue/appendicularia
....
|
|
/home/y/YourName
For small things. This is only suitable for small things such as configuration files or source code. Your home directory is limited up to 50GB.
/flash/UnitName/YourName
For computing outputs. We need to use /flash/XxxxU/ for any computing output. So, outputs file derived from sbatch jobs should be saved in /flash/XxxxU. These output files should be scp to your directory in the /bucket/XxxU directory.However, these outputs should be moved to /bucket/XxxxU soon.
/bucket/UnitName/YourName
For keeping your data. Sequence data and softwares can be placed at /bucket/UxxxU
Copy file
Use "scp" to copy data to bucket in your jobs. Deigo is read-only on the compute nodes. Instead we copy the data through the login nodes.
#!/bin/bash
#SBATCH --job-name=scp
#SBATCH --partition=compute
#SBATCH --mem=1G # 1G is used fo all cpus-per-task=2
#SBATCH --cpus-per-task=2 # max 24, corresponding to "-T 2"
#SBATCH --ntasks=1 # 1 task. Usually, 1
scp -r trinity_out_Doliolum3rd \
yourid@deigo.oist.jp:/bucket/XxxU/yourname/
More detail
Please see a page on research storage for more detail:
https://groups.oist.jp/scs/research-storage
(November 2020)
|
|
How to submit a job
Copy the following script as testjob.slurm, and type,
sbatch testjob.slurm
|
| testjob.slurm is as follows:
|
#!/bin/bash
#SBATCH --job-name=mp_raxml
#SBATCH --partition=compute
#SBATCH --mem=1G # 1G is used fo all cpus-per-task=2
#SBATCH --cpus-per-task=2 # max 24, corresponding to "-T 2"
#SBATCH --ntasks=1 # 1 task. Usually, 1
[Write the command line for your program.]
|
|
| cp
|
#!/bin/bash
#SBATCH --job-name=test
#SBATCH --partition=compute
#SBATCH --mem=1G # 1G is used fo all cpus-per-task=2
#SBATCH --cpus-per-task=2 # max 24, corresponding to "-T 2"
#SBATCH --ntasks=1 # 1 task. Usually, 1
cp testjob.slurm testjobCP.slurm
|
|
| MAFFT
|
sbatch the following file, job.slurm.
sbatch job.slurm
jobscript is as follows:
#!/bin/bash
#SBATCH --job-name=mafft
#SBATCH --partition=compute
#SBATCH --mem=2G
#SBATCH --cpus-per-task=2
#SBATCH --ntasks=1 # 1 task
mafft example2.txt > out_example2.txt
mafftDir.tar.gz
|
|
| BEAST2
|
#!/bin/bash
#SBATCH --job-name=job_script
#SBATCH --partition=compute
#SBATCH --mem=2G # It means total memory. "--mem-per-cpu=1G" is same meaning due to "--cpus-per-task=2"
#SBATCH --cpus-per-task=4 # max 24. It corresponds to "-threads 4"
#SBATCH --ntasks=1 # 1 task
~/bin/beast/bin/beast -threads 4 Sky.xml
|
|
| RAxML
|
sbatch the following file, job.slurm.
sbatch job.slurm
jobscript is as follows:
#!/bin/bash
#SBATCH --job-name=job_script
#SBATCH --partition=compute
#SBATCH --mem=2G # It means total memory. "--mem-per-cpu=1G" is same meaning due to "--cpus-per-task=2"
#SBATCH --cpus-per-task=2 # max 24. It corresponds to "-T 2"
#SBATCH --ntasks=1 # 1 task
raxmlHPC-PTHREADS-SSE3 -f a -x 12345 -p 12345 -# 100 -m GTRGAMMA -s Coe5_500.PHYLIP -q part -o Scca -n outfile -T 2
slurmRAxML_WebExample5spp.tar.gz
Note: -T 2 is faster than -T 4.
|
|
RAxML via array job
Make a file describing file list (010_param.txt) by 010_autoListMaker.pl.
ENSORLP00000001536.txt Chicken_ENSGALP00000015412_CDK6
ENSP00000013070.txt Drosophila_FBpp0080532_CG15141
ENSP00000013807.txt Drosophila_FBpp0086578_Ercc1
ENSP00000215882.txt SeaSquirt_ENSCINP00000025857_NONE
ENSP00000218516.txt Anole_ENSACAP00000005195_NONE
ENSP00000221114.txt Drosophila_FBpp0080701_l37Ce
ENSP00000222250.txt Drosophila_FBpp0291104_Vdup1
ENSP00000225609.txt Drosophila_FBpp0083251_CG4433
ENSP00000231668.txt Drosophila_FBpp0078315_CG2023
ENSP00000239461.txt SeaSquirt_ENSCINP00000019608_NON
....
Then sbatch the following file, 015_raxArraySLURM.slurm.
#!/bin/bash
#SBATCH --job-name=array_raxml
#SBATCH --partition=compute
#SBATCH --mem=2G # It means total memory. "--mem-per-cpu=1G" is same meaning due to "--cpus-per-task=2"
#SBATCH --cpus-per-task=2 # max 24. It corresponds to "-T 2"
#SBATCH --ntasks=1 # 1 task
#SBATCH --array=1-100%240 # 12*20 amounts to 5% of all nodes ## --array=5,15,15200 style is also acceptable
tid=$SLURM_ARRAY_TASK_ID
params="$(head -$tid 010_param.txt | tail -1)"
param1=${params% *} # Shell Parameter Expansion.Eng,Jap.
param2=${params#* }
./raxmlHPC-PTHREADS-SSE3 -f a -x 12345 -p 12345 -# 5 -m GTRGAMMA -s 010_sequenceFileDir/$param1 -q 010_partitionFileDir/$param1 -o $param2 -n $param1 -T 2
raxmlSLURMArray.tar.gz (April 2016)
|
|
| BLAST+ |
[cluster:slurmJobs]$ cat job.slurm
#!/bin/bash
#SBATCH --mail-user="jun.inoue@oist.jp"
#SBATCH --mail-type=FAIL,END
#SBATCH --mem=5G
#SBATCH -p compute
#SBATCH -a 1-3 # number of lines in list
#SBATCH --cpus-per-task=1
#SBATCH --time=00:00:10
i=$SLURM_ARRAY_TASK_ID
dir="/home/j/jun-inoue/onihitode/slurmJobs"
blast="/home/j/jun-inoue/bin/ncbi-blast-2.2.31+/bin"
pro="blastn"
query="queryAkajimaPheStart.txt"
list="list"
file="$(cat ${list} | sed ''${i}'!d' )"
out_dir="${pro}_q_dnr"
db_dir="/home/j/jun-inoue/onihitode/blastDB_454Scaffoldsfna"
db="${db_dir}/${file}_454Scaffolds.fna"
outfile="${pro}_${file}_dx.csv"
## This script can be checked by "sh job.slurm"
##
if you uncomment the following line.
#echo ${blast}/${pro} \
# -query ${query} \
# -db ${db} \
# -evalue 0.0001 -outfmt 6 -num_threads 12 -num_alignments 10 \
# \> ${out_dir}/${outfile}
cd ${dir}
mkdir ${out_dir}
${blast}/${pro} \
-query ${query} \
-db ${db} \
-evalue 0.0001 -outfmt 6 -num_threads 12 -num_alignments 10 \
> ${out_dir}/${outfile}
|
[cluster:slurmJobs]$ ls
blastn_q_dnr
job.slurm
queryAkajimaPheStart.txt
list
[cluster:slurmJobs]$ cat list
COTS-1_S1
COTS-3_S2
COTS-4_S3
[cluster:slurmJobs]$ ls ../blastDB_454Scaffoldsfna
COTS-1_S1_454Scaffolds.fna
COTS-1_S1_454Scaffolds.fna.nhd
COTS-1_S1_454Scaffolds.fna.nhi
COTS-1_S1_454Scaffolds.fna.nhr
COTS-1_S1_454Scaffolds.fna.nin
COTS-1_S1_454Scaffolds.fna.nog
COTS-1_S1_454Scaffolds.fna.nsd
COTS-1_S1_454Scaffolds.fna.nsi
COTS-1_S1_454Scaffolds.fna.nsq
COTS-3_S2_454Scaffolds.fna
....
[cluster:slurmJobs]$ ls blastn_q_dnr/
blastn_COTS-1_S1_dx.csv
blastn_COTS-3_S2_dx.csv
blastn_COTS-4_S3_dx.csv
|
|
| OrthoFinder (May 2021) |
#!/bin/bash
#SBATCH --job-name=orthofinder
#SBATCH --partition=compute
#SBATCH --time=1:00:00
#SBATCH --mem=10G
#SBATCH --cpus-per-task=8
#SBATCH --ntasks=1 # 1 task
NUM_SRD=$SLURM_CPUS_PER_TASK
#export PATH=$HOME/homeMCL/bin:$PATH
python3 orthofinder.py -f /flash/SatohU/inoue/OrthoFinder/ExampleData -s SpeciesTree.txt -t $NUM_SRD -y
|
|
test_yuraiParallel.tar.gz (73MB)
0. Prepare querySeqFiles dir.
1. Create list files 010_list*.txt.
perl 010_list0.txt
2. Run array job.
Outfiles will be saved in 020_outdir (automatically made).
Change the following line according to the number of list files.
#SBATCH --array=0-3
sbatch 020_arrayJobAA.slurm
(Aug. 2018)
|
|
|
sbatch
Submit a job script. See documentation of SchedMD.
squeue
Report the state of nodes managed by slurm.
squeue -u jun-inoue
Report the state of nodes for a specific user.
scancel
Cancel a running job step.
sbatch --help
Show information.
|
|
How to sbatch your job script
0. R script
Make a R script, test.R.
hel <- "hello"
write(hel, file="out.txt")
Method 1. Rscript
Save the following lines in a file, job.slurm, and sbatch it "sbatch job.slurm".
#!/bin/bash
#SBATCH --job-name=job_script
#SBATCH --partition=compute
#SBATCH --mem-per-cpu=1G
#SBATCH --ntasks=1
Rscript /home/j/jun-inoue/testSLURM_R/test.R
job script requiring longer time
#!/bin/bash
#SBATCH --job-name=Sn2
#SBATCH --partition=compute
#SBATCH --time=16:00:00
#SBATCH --mem-per-cpu=1G
#SBATCH --ntasks=1
Rscript /work/SinclairU/inoue/R_cal/Ens76/Sn2Dir/Tancha2.R
"#SBATCH --time=7-00:00:00 " means at most 7 days.
|
|
SRA Toolkit is availabe. To check available softwares on sango, type,
module avail
For module, see "Use the module system" page.
(Oct. 2018)
|
| Below indicates a job file for fastq-dump. fastq-dump downlods a paired-end file and make two files, XXXX_1 and XXXX_2. See this page (JPN). |
#!/bin/bash
#SBATCH --job-name=prefetch
#SBATCH --mail-user="jun.inoue@oist.jp"
#SBATCH --partition=compute
#SBATCH --mem=2G
#SBATCH --cpus-per-task=2
#SBATCH --ntasks=1 # 1 task
/apps/free/sra-tools/2.8.2-1/bin/fastq-dump SRR390728 --split-files
|
|
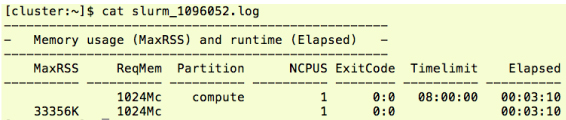 |
We can identify the required memory from the log file. Default setting of Timelimit is depending on the partition. See "SLURM partitions in Sango" for the partition information. See documentation of SchedMD. |
|