|
|||
| 2020 年 6 月 13 日 改訂 | |||
NOVOPlasty はミトコンドリアや葉緑体ゲノム専用のアセンブラです.
インファイル ヒトデ類のミトコンドリアゲノム解析の例です.
上記ファイルを test_config.txt として保存し,以下のコマンドで走らせます.
アウトファイル 脊椎動物のミトコンドリアゲノムなどでは,MitoAnnotator でアノテーションすると良いと思います.ヒトデ類などでも,だいたいうまくアノテーションされます.
Merged_contigs_*.txt
Contigs_1_*.fast このファイルに保存される contig が使える場合もあります. |
|||
|
|||
| Novoplasty_mtGenomePopulation.tar.gz 解析には,MAFFT, TrimAl, RAxML が必要です. |
|||
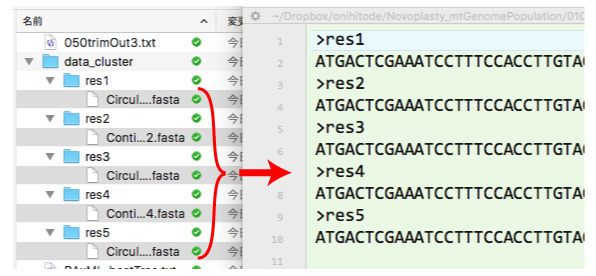
python3 010collectSeq.py |
|||
 |
|||
| 得られた結果は 010out.txt に保存されます.普通,010out.txt に並べられた mtGenome 配列 (環状) の先頭は,揃っていません.このため,Editor の検索機能を使って,配列の先頭を揃える必要があります. |
|||
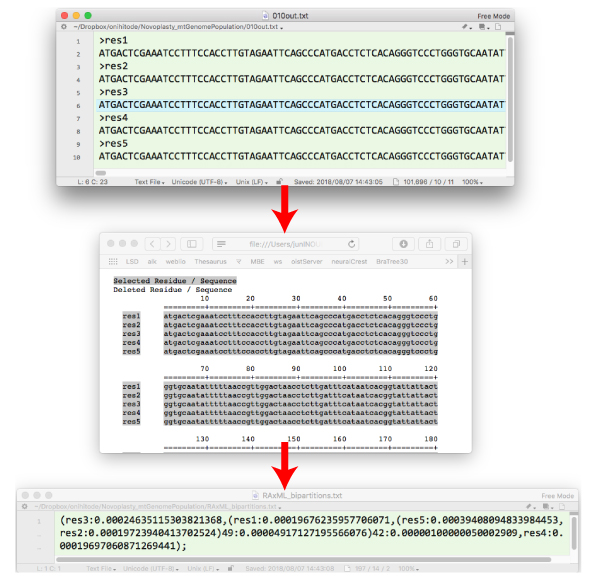
| sh 020command.sh | |||
 |
|||
| MAFFT でアライメントを行います.そのあと,trimAl で,unambiguously aligned sites を取り除きます.得られた配列を RAxML で解析します. |
|||
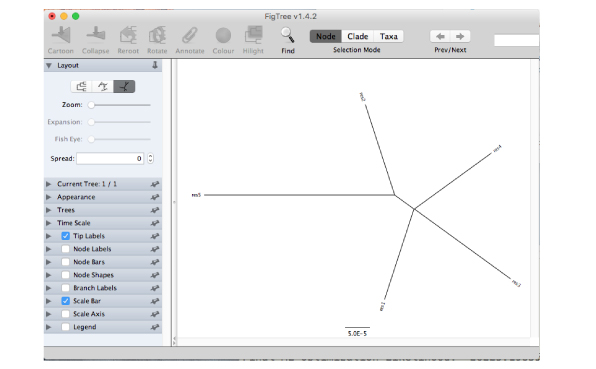
RAxML_bipartitions.txt を FigTree で開く. |
|||
 |
|||
| (2020 年 6 月) | |||
|
|