|
|
| 2012 年 11 月 30日 改訂 |
Bioperl は既に作成された perl のスクリプトをダウンロードして利用するシステムのことです.GenBank format から必要な情報を抜き出す等の作業ができます.Bioperl を使うには Perl の知識が必要で,簡単な参考書を一冊終わらせておく必要があります.
Web で得られるチュートリアルは,どれもあまりわかりやすいとは言えません.私の場合は,文法は完全にはわからないけれど,web 検索で出てきた BioPerl のスクリプトを切り貼りして print out などを書き換えて,何とか求めるスクリプトが完成する感じです.
Bioperl のスクリプトを用いて,web からたくさんのファイルをダウンロードする場合 (一晩ぐらいでも),大量データを処理している途中にストップすることがよくあります.もう一度同じことをすると,正常に作動することが多いです.web 通信の問題だと思っています.シェルスクリプトを作成して,同じ命令を何度か行うようにすると便利です.シェルスクリプトの作り方はこちらをご覧下さい.
2011 年 11 月の時点で,およそ 2 年間にわたって BioPerl をごくたまに使ってきましたが,正直なところ私のようなユーザーに BioPerl はあまり向いていません.Perl の利点はスクリプトを自分なりに書き換えて便利にする点にあると思います.BioPerl のモジュール群から目的にあったプログラムを探すのは簡単とは思えないですし,BioPerl のスクリプトを書き換えるにはかなり Perl の知識が必要です.Perl のスクリプトは自分ですぐに変更できないとあまり意味が無いと思います.
|
|
セットアップ
CPAN (Comprehensive Perl Archive Network) は,Perl のスクリプトを保管しているネットワークです.実際には,perl のモジュールをダウンロードして利用するシステムのことです.BioPerl を使うには CPAN を簡単にでも使える必要があります.CPAN はインターネットにつながっていないと使えません.
CPAN シェルを呼び出すのは,
sudo CPAN
です.パスワードを聞いてくるので答えましょう.
だめなときは,
sudo perl -MCPAN -e shell
としてとすれば,CPAN シェルのプロンプト「cpan>」が現れます.
出るときは「quit」あるいは「q」です.
「sudo CPAN」がうまく行かないときは,二つ目のコマンドで cpan シェルにした後で,「install Bundle::CPAN」とすれば,CPAN モジュールがインストールされます.
以上は,こちらに従って操作を行いました.CPAN の最初の段階としてこのサイトはとても有用です.
ウェブサイト
CPAN のウェブサイトを直接見に行くことができます.こちらです.
このウェブサイトでは主にモジュールの使い方を調べます.インストールは以下に示した CPAN シェルを使った方が楽です. 例えば,calendar などで検索すると,こちらにたどり着きます.右はじにある Download: から tarball (.tar.gz ファイル) をダウンロード&解凍して README を開いてください. ここに書いてある BASIC USAGE は熟読した方が良いそうです.
|
|
|
インストールの手順
BioPerl への道に従ってインストールしました.
perl -MCPAN -e "force install Bundle::BioPerl"
あるいは sudo をつけて,
sudo perl -MCPAN -e "force install Bundle::BioPerl"
でインストールが始まるはずです.うまく始まると,インストールの終了に 15 分ぐらいかかります.いろいろ聞かれますが,すべてリターンを押して yes を選びます.
最近になって perl を 5.12 にアップグレードした (インストールし直しました) ところ,BioPerl が使えなくなりました.このため BioPerl もインストールし直しました.最初上記のコマンドをうったところ,
Alert: While trying to 'parse' YAML file
.....
というエラーメッセージが出ました.こちらのページに従っていろいろやっているうちに,自分でも理解できませんが上記のコマンドが走るようになりました [2011 年 11 月].
確認
環境が整ったかどうかは,
perl -MBio::Perl -le 'print $Bio::Perl::VERSION'
とうつと,1.006001 のような数字が返っています.[しかし,2012 年 12 月になってインストール後にこのコマンドを打ったら,何も返事がありませんでした.でもインストールは成功していました.]
あるいはこちら (BioPerl_test.tar.gz) のプログラムをダウンロードしてテストしてください.Bptutorial.pl の最初に出てくる例題です (一部変更).GenBank といタンパク質データバンクからダウンロードするので,インターネットに PC がつながっている必要があります.「perl test.pl」と入力してください.Fasta 形式の配列が「AB005035.fas」というファイル名で保存されます.
ダウンロード&解凍して得られたディレクトリに入り,
perl test.pl
と入力して下さい..fas という拡張子を持つファイルが得られ,ファスタ形式の形式が保存されます.
use Bio::Perl;
# please type,
# perl test.pl
# this script will only work if you have an internet connection on the
# computer you're using, the databases you can get sequences from
# are 'swiss', 'genbank', 'genpept', 'embl', and 'refseq'
$seq_object = get_sequence(genbank,"AB005035");
write_sequence(">AB005035",'fasta',$seq_object);
データベースに求める配列が無いと,以下のようなメッセージが出ます.このときは,ウェブなどでアクセッション番号等を確かめましょう.
------------- EXCEPTION: Bio::Root::Exception -------------
MSG: id does not exist
STACK: Error::throw
STACK: Bio::Root::Root::throw /Users/inouejun/src/bioperl-1.2.3/Bio/Root/Root.pm:342
STACK: Bio::DB::WebDBSeqI::get_Seq_by_id /Users/inouejun/src/bioperl-1.2.3/Bio/DB/WebDBSeqI.pm:155
STACK: Bio::Perl::get_sequence /Users/inouejun/src/bioperl-1.2.3/Bio/Perl.pm:513
STACK: 4-1.pl:2
-----------------------------------------------------------
失敗例
久しぶりに BioPerl を動かしてみたら,以下のようなメッセージが出ました.Perl のバージョンが古いとまずいようです.Perl と BioPerl を最新のものに入れ替えたら,無事動くようになりました.入れ替えには 15 分ほどかかりました.
[inouejun:270_NutRelParser2_fol]$ 010_DupNodeLister.pl
Can't locate Bio/TreeIO.pm in @INC (@INC contains: /sw/lib/perl5 .... /System/Library/Perl/Extras/5.10.0 .) at ./010_DupNodeLister.pl line 6.
BEGIN failed--compilation aborted at ./010_DupNodeLister.pl line 6.
|
|
追加
BioPerl で書かれたスクリプトを動かしたら,以下のようなエラーメッセージが出ることがあります.この場合は,見つからないモジュールをインストールする必要があります.エラーメッセージとスクリプトを見ると,「Bio::TreeIO」をインストールする必要があります.
[inouejun:270_NutRelParser2_fol]$ 010_DupNodeLister.pl
Can't locate Bio/TreeIO.pm in @INC (@INC contains: /sw/lib/perl5 .....) at ./010_DupNodeLister.pl line 6.
BEGIN failed--compilation aborted at ./010_DupNodeLister.pl line 6.
sudo CPAN で CPAN を立ち上げます.
[inouejun:270_NutRelParser2_fol]$ sudo CPAN
Password:
Terminal does not support AddHistory.
cpan shell -- CPAN exploration and modules installation (v1.9456)
Enter 'h' for help.
install Bio::TreeIO と打ち込むと,インストールが始まります.
cpan[1]> install Bio::TreeIO
確認
こちらを参照しました.Web::Scraper というモジュールを CPAN からインストールした後に確認しています.
CPAN シェルから,以下のコマンドをうちます.
cpan[2]> i /Web::Scraper/
Module < WWW::Mechanize::Plugin::Web::Scraper (BLOM/WWW-Mechanize-Plugin-Web-Scraper-0.02.tar.gz)
Module = Web::Scraper (MIYAGAWA/Web-Scraper-0.37.tar.gz)
Module < Web::Scraper::Config (DMAKI/Web-Scraper-Config-0.01.tar.gz)
Module = Web::Scraper::Filter (MIYAGAWA/Web-Scraper-0.37.tar.gz)
Module = Web::Scraper::LibXML (MIYAGAWA/Web-Scraper-0.37.tar.gz)
5 items found
モジュールがインストールされている場所は,以下で確認できます.
[JunINOUE:~]$ perldoc5.12 -ml Web::Scraper
/Library/Perl/5.12/Web/Scraper.pm
|
モジュールのマニュアルインストール (例: StandAloneFasta ) |
|
マニュアルインストールとは
CPAN などを用いたモジュールのインストールがなぜかうまく行かない場合は,手動でインストールしてみるとよいことがあります.今回は,私が何とか解決したトラブルを例にとって,マニュアルインストールを説明します.
背景
友人が書いた fasta_XX.pl が,うまく動きません.
[inouejun:270_NutRelParser2_fol]$ ./fasta_XX.pl test.ret lizard_ORg.conv
Can't locate Bio/Tools/Run/Alignment/StandAloneFasta.pm in @INC (@INC contains: /sw/lib/perl5 /sw/lib/perl5/darwin .....) at ./fasta_OR.pl line 15.
BEGIN failed--compilation aborted at ./fasta_OR.pl line 15.
このようなメッセージが得られたとします.太字のメッセージが返ってきたので,CPAN から以下によって該当するモジュールをインストールしました.
install Bio::Tools::Run::Alignment::StandAloneFasta
しかし,CPAN から 「cpan[2]> i /Bio::Tools::Run::Alignment::StandAloneFasta/」としてインストールが完了したか確認しましたが,どうもうまくできていないようです.
cpan[2]> i /Bio::Tools::Run::Alignment::StandAloneFasta/
Module id = Bio::Tools::Run::Alignment::StandAloneFasta
CPAN_USERID CJFIELDS (Christopher Fields <cjfields@bioperl.org>)
CPAN_FILE C/CJ/CJFIELDS/BioPerl-Run-1.006900.tar.gz
UPLOAD_DATE 2011-04-21
MANPAGE Bio::Tools::Run::Alignment::StandAloneFasta - Object for the local execution of the Fasta3 programs ((t)fasta3, (t)fastx3, (t)fasty3 ssearch3)
INST_FILE (not installed)
そこで,マニュアルインストールを行いました.
マニュアルインストール
今回のマニュアルインストールは MacPorts というパッケージシステムのインストールから始めています.その大きな理由は,私が経験した以下の問題点を回避する点にあったと今思います.
- 現在使っている perl (which perl で検索) がインストールされている場所が PATH で適切に指定されていない.
- StandAloneFasta モジュールをインストール場所が,現在使われている perl と対応していない (どこにマニュアルインストールしたら良いかわからない).
上記の問題は,CPAN 経由のモジュールを使い慣れている方に取っては簡単なことなのでしょう.MacPorts は,OSX では人気のあるシステムのようなので,今後調べてゆこうと思います.今回はとりあえずインストールしました.
1.
http://poccori.com/blog/wp/archives/90
に従って,MacPorts をインストールしました.
MacPorts をインストールした後は,以下によってアップデートを行う必要があるようです.
$ sudo port -d selfupdate
$ sudo port -d sync
2.
上記のページに従い,perl5 や vim に加えて一通り例題のもの (wget や subversion, nkf など) をインストールしました.
~/.bash_profile に以下の環境変数を追加するのを忘れないでください.
export PATH=/opt/local/bin:/opt/local/sbin/:$PATH
export MANPATH=/opt/local/man:$MANPATH
Perl がインストールされたかどうか確認しましょう.
[inouejun:bin]$ which perl
/opt/local/bin/perl
3.
http://search.cpan.org/~cjfields/BioPerl-Run-1.006900/lib/Bio/Tools/Run/Alignment/StandAloneFasta.pm
の右側にある BioPerl-Run-1.006900.tar.gz をダウンロード&解凍しました.
4.
http://poccori.com/blog/wp/archives/90
「手動でインストールを行いたい場合 (CPANシェルをあえて使わない場合)」
にしたがって,解凍したファイルを /opt/local/lib/perl5/5.12.4 にコピーしました.
5.
コピーしたファイルである /opt/local/lib/perl5/5.12.4/BioPerl-Run-1.006900 に入ります.
6.
あとは INSTALL の MANUAL INSTALLATION を行いました.
> sudo perl Build.PL
> sudo ./Build test
> sudo ./Build install
テストはエラー?が出たようですが,気にしませんでした.もしかしたらあとで問題になるかもしれません.
上の操作は Makefile.PL がなく,Build.PL がある場合です.この場合は Module::Build ベースのディストリビューションだそうです.多くのモジュールは,以下のような Make.PL を用いた,以下の手順で手動インストールを行います.
> perl Makerile.PL
> make
> make test
> sudo make install
7.
最後は以下のコマンドでインストールされた場所を確認しました.
[JunINOUE:BioPerl-Run-1.006900]$ perldoc -ml Bio::Tools::Run::Alignment::StandAloneFasta
/opt/local/lib/perl5/5.12.4/BioPerl-Run-1.006900/blib/lib/Bio/Tools/Run/Alignment/StandAloneFasta.pm
あるいは,cpan をつかっても,インストールされたかどうかを確認することができます
cpan[1]> i /Bio::Tools::Run::Alignment::StandAloneFasta/
Going to read '/Users/JunINOUE/.cpan/Metadata'
Database was generated on Fri, 30 Nov 2012 00:31:03 GMT
Module id = Bio::Tools::Run::Alignment::StandAloneFasta
CPAN_USERID CJFIELDS (Christopher Fields <cjfields@bioperl.org>)
CPAN_FILE C/CJ/CJFIELDS/BioPerl-Run-1.006900.tar.gz
UPLOAD_DATE 2011-04-21
MANPAGE Bio::Tools::Run::Alignment::StandAloneFasta - Object for the local execution of the Fasta3 programs ((t)fasta3, (t)fastx3, (t)fasty3 ssearch3)
INST_FILE /opt/local/lib/perl5/site_perl/5.12.4/Bio/Tools/Run/Alignment/StandAloneFasta.pm
INST_VERSION undef
|
|
|
モジュールについて調べるには,以下のようなコマンドをうちます.
perldoc Bio::DB::GenBank
その後,
/Seq_by_acc
のようにタイプし,必要な部分を検索します.ただ,ここにすべてのことが書いてある訳ではありません.その場合は,google 検索で調べるか,あるいは,GenBank.pm を開けてみてコメント等を読んでみた方が良いことが多いです.GenBank.pm に無くても,その中で他のモジュールを使っていることもあるので,さらにそれも見てみると良いでしょう.モジュールの場所がわからないときは,ホームディレクトリから 「find . -name GenBank.pm」などと打つと,最初の方に出てきます.時間がかかるので,検索が最後まで終わるのを待つ必要は無いです.
|
|
|
HOWTO:Beginners にある例題です.以下をコピーして seqio.pl という名前で保存し,ターミナルで同じフォルダに入って「perl seqio.pl」と入力してください.
#!/bin/perl -w
# 単に配列をスクリーンにプリントアウトします.
use Bio::Seq; # Seq.pm という名前のモジュールです.Bio/Seq.pm に保存されています.
$seq_obj = Bio::Seq -> new(-seq => "aaaatgggggggggggccccgtt",
-display_id => "#12345",
-desc => "example 1",
-alphabet => "dna" );
print $seq_obj -> seq(),"\n";
#!/bin/perl -w
# 配列を GenBank 形式で sequence.gb というファイルとして保存します.
use Bio::Seq;
use Bio::SeqIO; # インプットとアウトプットを行うモジュールです.
$seq_obj = Bio::Seq -> new(-seq => "aaaatgggggggggggccccgtt",
-display_id => "#12345",
-desc => "example 1",
-alphabet => "dna" );
$seqio_obj = Bio::SeqIO -> new(-file => '>sequence.gb', -format=>'genbank');
# fasta, EMBL, SwissProt などでも保存できます.詳しくは perldoc Bio::SeqIO をタイプして -format の説明を読むか,google で Bio::SeqIO で検索して,こちらのようなサイトを見つけてください.
$seqio_obj -> write_seq($seq_obj);
|
|
taxon ID を調べることで,スペルチェックをします.きっとより良い方法があるので,今後改善してゆきます [2011 年 1 月].
アウトプットはこんな感じです.ボールドのスペルにミスがあります.
[inouejun:spellCheckSpName_fol]$ spellCheckSpName.pl
SpName taxonID
Branchiostoma belcheri 7741
Use of uninitialized value $taxonid in concatenation
(.) or string at ./spellCheckSpName.pl line 18.
Epstatretus burgeri
Lethenteron reissneri 7753
スクリプトです.
#!/usr/local/bin/perl
use strict;
use warnings;
use Bio::DB::Taxonomy;
my $db = new Bio::DB::Taxonomy(-source => 'entrez');
#my $spName = "Homo sapiens";
open(INFILE,"Names.txt");
my @inFile = <INFILE>;
close INFILE;
print "SpName taxonID\n";
foreach my $spName (@inFile) {
chomp($spName);
my $taxonid = $db->get_taxonid($spName);
print "$spName $taxonid\n";
}
spellCheckSpName_fol.tar.gz
|
|
|
Fasta 形式の配列を読み込んで,スクリーンアウトにプリントします.
#!/bin/perl -w
use Bio::SeqIO;
use Bio::Perl;
$seqio_obj = Bio::SeqIO -> new(-file => "3sequence.fasta", -format => "fasta" );
# 配列が一つだけの場合.
$seq_obj = $seqio_obj -> next_seq;
print $seq_obj -> seq()."\n";
# 配列が複数は行っている場合 (上の 2 行をコメントアウトし,下 3 行の # を取り除いてください)
#while ($seq_obj = $seqio_obj->next_seq){
#print $seq_obj->seq,"\n";
#}
SeqFromFile_fol.tar.gz
|
| Fasta 形式で保存した塩基配列を翻訳する - multiple sequences |
|
mtDNA: 例題
while の部分は「perldoc use Bio::SeqIO;」を参照しました.
他の部分は こちらを参照しました.
「-codontable_id」で指定されている遺伝暗号については こちらをご覧下さい.
#!/usr/bin/perl -w
# perl NucTranslater_mt.pl test.fas
use Bio::Perl;
use Bio::SeqIO;
$inFile = "$ARGV[0]";
$outFile = $inFile.".prot";
$in = Bio::SeqIO -> new('-file' => "$inFile", '-format' => "Fasta");
while ($seq = $in->next_seq()) {
$translate = $seq->translate(-codontable_id => 2);
write_sequence(">>$outFile",'fasta',$translate);
}
|
| Fasta 形式で保存した塩基配列を翻訳する - 1 sequence |
|
3 種類の開始地点から翻訳した結果を返します.
核 DNA: 例題
mtDNA: 例題
#!/usr/bin/perl -w
# perl NucTranslater.pl 78644093.txt
use Bio::Perl;
use Bio::SeqIO;
$file = shift;
$in = Bio::SeqIO -> new('-file' => "$file", '-format' => "Fasta");
$seq = $in->next_seq();
print "\nNucleotide:\n",$seq->seq(),"\n";
$translate = $seq->translate;
print "\nFrame1:\n" ,$translate->seq();
$translate = $seq->translate(undef,undef,1);
print "\n\nFrame2\n: " ,$translate->seq();
$translate = $seq->translate(undef,undef,2);
print "\n\nFrame3\n: " ,$translate->seq(), "\n\n"
|
| Accession number から塩基配列を得る |
|
#!/usr/bin/perl -w
# FasHashListed.pl seq.fas list
use Bio::Perl;
$test_seq = get_sequence('genbank', "AB005035");
write_sequence(">AB005035.fasta",'fasta',$test_seq);
ここに示すように,GenBank を指定していますが,accession number は DDBJ のものでも問題なく配列を得ることができます.
010_SequenceRetriever.pl.tar.gz
こちらにある例題を参考にしました.
|
| Accession number から GenBank format を得る |
|
|
アミノ酸配列のGenBank フォーマットを得ます.
#!/usr/bin/perl -w
use Bio::DB::GenBank; # 他にも SwissProt (Bio::DB::SwissProt) や EMBL (Bio::DB::EMBL) などがあります.くわしくはこちらを参照してください.
use Bio::SeqIO;
$db_obj = Bio::DB::GenBank -> new;
$seq_obj = $db_obj -> get_Seq_by_id("68270954");
# get_Seq_by_acc (AAY88974) だと,AAY88974 という accession number を指定します.
$seqio_obj = Bio::SeqIO -> new(-file => '>gi68270954.gbk', -format => 'genbank' );
# 上記のように fasta, EMBL などでも保存できます.
$seqio_obj -> write_seq($seq_obj);
* get_Seq_by_acc の部分は 68270954 のような gi number でも,AAY88974.1 のような GenBank の number でも得られる結果は同じみたいです.
|
| Accession number から塩基配列を得てアミノ酸に翻訳する |
|
アミノ酸の GenBank format が得られた場合に,塩基配列の Accession number と CDS の位置が明示されていれば,以下のコードで CDS を得てタンパク質に翻訳します.3 種類の開始点を試しています.開始位置を確認することができます.こちらを参考にさせていただきました.
#!/usr/bin/perl -w
use Bio::Perl;
$AccNum_Nuc = "AM084879.1";
$range_start = "1";
$range_end = "663";
$test_dna = get_sequence('genbank', "$AccNum_Nuc");
$short_dna = $test_dna->trunc($range_start,$range_end);
print "\nNuc_AccNumber: $AccNum_Nuc\n\nCDS:\n";
print $short_dna->seq(),"\n\nTranslates to:";
$translate = $short_dna->translate;
print "\nFrame1: " ,$translate->seq();
$translate = $short_dna->translate(undef,undef,1);
print "\n\nFrame2: " ,$translate->seq();
$translate = $short_dna->translate(undef,undef,2);
print "\n\nFrame3: " ,$translate->seq(), "\n\n"
NucAccToCdsTrans.pl.tar.gz
結果はこんな感じです.
Nuc_AccNumber: AM084879.1
CDS:
CTCCTGGAGGTCATTGAGCCAGAGGTGTTATATTC
Translates to:
Frame1: LLEVIEPEVLYSGYDSSMPDTTWRIMS
Frame2: SWRSLSQRCYIRDMTVQCLIPLGGSCQ
Frame3: PGGH*ARGVIFGI*QFNA*YHLEDHVN
|
| GI mumber から accession number を得る |
|
論文には gi number よりもとの accession number を載せるようです.gi number から accession number を得るスクリプトを書きました.以下のようなアウトプットが得られます.
46849374 AB111383.1
BC045218 BC045218.1
AB111376 AB111376.1
120408644 EH284100.1
こちらの「Get accessions (actually accession.versions) for a list of GenBank IDs (GIs)」を参考にしました [2011 年 1 月].
ginumToaccnum_fol.tar.gz
|
|
Monodelphis_domestica の EST 配列をすべてダウンロードします.キーワードの書き方はこちらを参照してください.
use Bio::DB::GenBank;
use Bio::DB::Query::GenBank;
$query = "Monodelphis_domestica[ORGN]";
# 以下のように遺伝子の名前と長さでしぼることもできます.
#$query = "Monodelphis_domestica[ORGN] AND topoisomerase[TITL] and 0:3000[SLEN]";
$query_obj = Bio::DB::Query::GenBank -> new(-db => 'nucest', -query => $query );
# -db は protein, nuccore, nucgss,nucest など NCBI にあれば使えるようです.
$gb_obj = Bio::DB::GenBank -> new;
$stream_obj = $gb_obj -> get_Stream_by_query($query_obj);
while ($seq_obj = $stream_obj -> next_seq) {
$seqio_obj = Bio::SeqIO -> new(-file => '>>Monodelphis_domestica_EST.fas', -format => 'fasta' );
# 上記のように genbank, EMBL などでも保存できます.
## Accession number と長さを調べることもできます.実際に NCBI の website に行って,同じ件数が得られるか確認できます.
# print $seq_obj -> display_id, "\t", $seq_obj -> length, "\n";
$seqio_obj -> write_seq($seq_obj);
}
|
| Fasta 形式を GenBank 形式に変更する |
|
|
fasta 形式で書かれたファイルを genbank 形式 に変更します.こちらの Transforming sequence files (SeqIO) を参考にしました.
#!/usr/bin/perl -w
use Bio::SeqIO;
$in = Bio::SeqIO->new(-file => "3out.fasta",
-format => 'Fasta');
$out = Bio::SeqIO->new(-file => ">3out.gb",
-format => 'genbank');
while ( my $seq = $in->next_seq() ) {$out->write_seq($seq); }
FastaToGenBank.tar.gz
|
|
|
#!/usr/bin/perl -w
use Bio::DB::GenBank;
use Bio::Species;
$db_obj = Bio::DB::GenBank->new;
$seq_obj = $db_obj->get_Seq_by_acc("68270954");
print "display_id\t", $seq_obj->display_id, "\n";
print "desc\t", $seq_obj->desc, "\n";
print "display_name\t", $seq_obj->display_name, "\n";
print "accession\t", $seq_obj->accession, "\n";
print "primary_id\t", $seq_obj->primary_id, "\n";
print "seq_version\t", $seq_obj->seq_version, "\n";
print "keywords\t", $seq_obj->keywords, "\n";
print "length\t", $seq_obj->length, "\n";
my $species_obj = $seq_obj->species;
print "species\t", $species_obj->binomial(), "\n";
print "seq\t", $seq_obj->seq, "\n";
|
|
NCBI の Nucleotide, Nucleotide EST, Protein, データベースから gi numbers をダウンロードするスクリプトです.gi number は local blast である種の配列だけを対象に検索したい場合に用います.以下の例題は "Anguilla japinica" を例にしています.このウェブサイトと同じ数の gi numbers が得られているはずです.
giRetriever.pl.tar.gz
# こちらを参考にしました.1, 2
[2010 年 12 月]
|
| タンパク質コード遺伝子から全ての転写領域の配列を得る |
|
タンパク質コーディング遺伝子の Gi number (塩基配列) から転写領域 (CDS) を切り出します. SplicedNucSeqRevBP.pl.tar.gz.
詳しくは, こちらの「How do I get the complete spliced nucleotide sequence from the CDS section?」をご覧下さい.
#!/usr/bin/perl -w
# How do I get the complete spliced nucleotide sequence from the CDS section?
# http://www.bioperl.org/wiki/FAQ
use Bio::DB::GenBank;
use Bio::Perl;
# use Bio::SeqIO;
# use Bio::Location::SplitLocationI
my $ginum = "886903";
$db_obj = Bio::DB::GenBank->new;
$seq_obj = $db_obj->get_Seq_by_gi("$ginum");
foreach my $feat ( $seq_obj->top_SeqFeatures ) {
if ( $feat->primary_tag eq 'CDS' ) {
my $cds_obj = $feat->spliced_seq;
print ">",$feat->gff_string,"\n",$cds_obj->seq,"\n";
}
}
結果です.> の行には転写されたアミノ酸配列などの情報を含めています.
>X82579 EMBL/GenBank/SwissProt
CDS 66 1271 . + . codon_start 1 ;
db_xref "GI:886904" "GOA:P51856"
"InterPro:IPR001664" "InterPro:IPR002957"
"InterPro:IPR011000" "UniProtKB/Swiss-Prot:P51856" ;
gene KRT19 ; product "keratin 19" ;
protein_id "CAA57915.1" ;
translation MTSYSYRQSSSTSTLGLG.....
ATGACCTCCTACAGCTACCGC.....
複数の GenBank ファイルから gi number を得て,ファイルごとの転写領域を得るスクリプトです.NucSplicer_fol.tar.gz.
|
| Newick 形式: 各 clade の OTU をリストアップする |
|
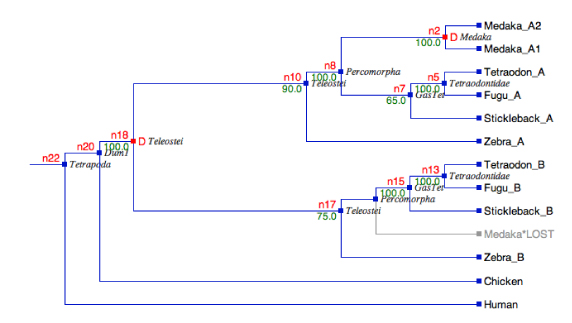
LeafPicker.pl tree.newick n18 > out_nwk
tree.newick という newick フォーマットで書かれた tree を処理します.n18 という名前の node 直下の n10 と n17 というそれぞれのクレードに含まれている OTU をリストアップします.こちらを参考にして作りました.ターミナルで「perldoc Bio::TreeIO」と打つと,多少参考になる情報が出てきます.
LeafPicker_fol.tar.gz
例題の tree.nhx を Notung で見ると,このようになっています.
アウトプットは以下です.
n18 each_Descendent
n10
n17
n18 get_all_Descendents
n10
n8
n2
Medaka_A1
Medaka_A2
n7
n5
Fugu_A
Tetraodon_A
Stickleback_A
Zebra_A
n17
r48
n15
n13
Fugu_B
Tetraodon_B
Stickleback_B
Medaka*LOST
Zebra_B
|
| Newick 形式: Lineage ごとの樹長を計算する |
|
こちらをご覧下さい.
|
|
Bio::Tree::TreeI を使うと,OTU 間の樹長を計算したり,ある種によって root を取り直したりできます.こちらをご覧下さい.
|
|
|
|
|
|
足りなりモジュールををインストール
例えば以下のようなメッセージが得られることがあります.このメッセージは大量のスクリーンアウトを上から見てゆくと,最初に出てくるエラーメッセージです.
......
CPAN: File::Temp loaded ok (v0.18)
Warning (usually harmless): 'YAML' not installed, will not store persistent state
......
ここでは,CPAN で「install YAML」 とすると,問題が解決することがあります.
|
|